

Evidence for Brain Glucose Dysregulation
in Alzheimer's diseaseThis section is compiled by Frank M. Painter, D.C.
Send all comments or additions to: Frankp@chiro.org


FROM: Alzheimers Dement. 2018 (Mar); 14 (3): 318–329 ~ FULL TEXT
Yang An, Vijay R. Varma, Sudhir Varma, Ramon Casanova, Eric Dammer et al.
Laboratory of Behavioral Neuroscience,
National Institute on Aging (NIA),
National Institutes of Health (NIH),
Baltimore, MD, USA.INTRODUCTION: It is unclear whether abnormalities in brain glucose homeostasis are associated with Alzheimer's disease (AD) pathogenesis.
METHODS: Within the autopsy cohort of the Baltimore Longitudinal Study of Aging, we measured brain glucose concentration and assessed the ratios of the glycolytic amino acids, serine, glycine, and alanine to glucose. We also quantified protein levels of the neuronal (GLUT3) and astrocytic (GLUT1) glucose transporters. Finally, we assessed the relationships between plasma glucose measured before death and brain tissue glucose.
RESULTS: Higher brain tissue glucose concentration, reduced glycolytic flux, and lower GLUT3 are related to severity of AD pathology and the expression of AD symptoms. Longitudinal increases in fasting plasma glucose levels are associated with higher brain tissue glucose concentrations.
There' more info like this in our: Nutrition Section DISCUSSION: Impaired glucose metabolism due to reduced glycolytic flux may be intrinsic to AD pathogenesis. Abnormalities in brain glucose homeostasis may begin several years before the onset of clinical symptoms.
Keywords: Glucose; Insulin resistance; Alzheimer’s disease; GLUT3; GLUT1; Neuritic plaque; Neurofibrillary tangles; Mass spectrometry; Glycolysis
From the Full Text Article:
Introduction
Although numerous epidemiological studies indicate that peripheral insulin resistance and diabetes are risk factors for Alzheimer’s disease (AD) [1–3], it is not known whether brain glucose dysregulation is a key feature of AD and is related to severity of AD pathology or symptom expression. [4, 5] Previous studies have shown that several components of the insulin signaling pathway are abnormal in AD brains relative to controls, including genes encoding insulin, IGF-1, and IGF-2 peptides and their receptors. [6–10] Because these abnormalities appear to be a common feature of both type-1 and type-2 diabetes, the term “type-3 diabetes” was proposed to describe brainspecific abnormalities in insulin signaling associated with AD. [11, 12] Taken together, the large body of evidence implicating abnormal insulin signaling in AD has led to clinical trials targeting these abnormalities in patients with mild cognitive impairment and AD. [13–15] However, it is well recognized that glucose transport from the peripheral circulation across the blood-brain barrier and capillary endothelial cells into the interstitial fluid and brain tissue are largely insulin-independent processes. [16, 17] Similarly, the transport of glucose across the cell membrane into neurons is largely independent of insulin [18]. Although 18F-deoxyglucose positron emission tomography (18FDGPET) studies have shown reduced brain glucose uptake in regions vulnerable to AD pathology [19–22], it is unclear whether an overall failure of regulation of brain glucose metabolism is a key etiopathogenic factor in AD and whether abnormalities of brain glucose homeostasis in AD are related to peripheral glucose concentration. Answering these questions is critical to establishing whether central glucose homeostasis is a potential target for diseasemodifying treatments in AD.
In this study, we asked the following main questions:
Is brain tissue glucose concentration altered in AD?
What is the relationship between brain tissue glucose concentration and severity of AD pathology?
What are plausible molecular mechanisms underlying abnormalities of brain glucose homeostasis in AD?
What is the relationship between trajectories of blood glucose concentration during life and brain tissue glucose levels measured at death?
Our results provide the first evidence for brain glucose dysregulation as a critical event in AD pathogenesis that closely reflects both severity of AD pathology and the expression of symptoms.
Methods
Participants
The Baltimore Longitudinal Study of Aging (BLSA) is a prospective, ongoing cohort study of community-dwelling volunteer participants in Baltimore that began in 1958 and has been described in detail previously [23, 24]. Historically, participants underwent extensive biomedical examination and neuropsychological testing every 2 years. From 2003, participants under age 60 years are assessed every 4 years; those aged 60 to 79 years every 2 years and participants aged 80 years and older are assessed annually. Written informed consent was obtained at each visit, and the study was approved by the local institutional review board and the National Institute on Aging. The participants in this report were from the autopsy program of the BLSA that was initiated in 1986 and has been described previously [25]. They provided data on concentrations of brain tissue glucose and the glycolytic amino acids, serine, glycine, and alanine (N = 43; from the middle frontal gyrus [MFG], inferior temporal gyrus [ITG], and cerebellum), as well as proteomic data from the MFG (N = 47). The mean age at death in the sample was 86.6 ± 9.5 years (range 62.9–99.2). As reported previously, the autopsy subsample is not significantly different from the BLSA cohort as a whole in terms of the rates of dementia and clinical stroke [26].
Cognitive status
At each assessment, participants underwent a battery of neuropsychological testing. Clinical and neuropsychological data were reviewed at consensus case conferences if they made four or more errors on the Blessed Information Memory Concentration test, if their Clinical Dementia Rating score was equal to or greater than 0.5, or if concerns were raised about their cognitive status. In addition, all participants were evaluated by case conference on death or withdrawal. The diagnoses of dementia and AD were based on the Diagnostic and Statistical Manual-III-R [27] and the National Institute of Neurological and Communication Disorders and Stroke–Alzheimer’s Disease and Related Disorders Association criteria [28], respectively.
Neuropathological studies
Postmortem brain examinations were performed by an experienced neuropathologist (J.C.T.). Assessment of neuritic plaques and neurofibrillary tangles using Consortiumto Establish a Registry for Alzheimer’s Disease (CERAD) [29] and Braak criteria [30], respectively, have been described previously [31]. We have previously described the clinico-pathological features of BLSA participants categorized as “asymptomatic Alzheimer’s disease (ASYMAD)” after neuropathological assessment at death [32]. Briefly, these individuals have significant AD neuropathology at autopsy, but without evidence for cognitive impairment during life, as assessed by longitudinal cognitive evaluations during their BLSA research visits.
Plasma glucose measurement
Plasma glucose measurements were obtained from venous blood samples after an overnight fast by the glucose-oxidase method as described previously [33]. We used all available longitudinal plasma glucose data (445 observations, mean follow-up interval. 19.1 years). We excluded 10 data points where fasting plasma glucose values were beyond three standard deviations from the mean value.
Quantitative metabolomics assays of brain tissue glucose and
glycolytic amino acids (serine, glycine, and alanine)
Glucose concentration was measured in frozen brain tissue samples from 43 BLSA participants (N = 14 AD; N = 14 control; and N = 15 “ASYMAD”) on the BIOCRATES P180 platform. Brain tissue regions for glucose assays were selected a priori in the MFG and ITG to represent brain regions vulnerable to amyloid and tau deposition, respectively. The cerebellum was sampled to represent an additional brain region resistant to classical AD pathology [34].
A sterile 4-mm diameter tissue punch was extracted from the cortical surface of the three brain regions, that is, MFG, ITG, and the cerebellum from brain tissue samples stored at –80°C. To extract metabolites from the brain tissue, samples were homogenized using Precellys with ethanol phosphate buffer. Samples were centrifuged, and the supernatant was used for analysis. The BIOCRATES P180 platform was used for the quantification of glucose and the three glycolytic amino acids, serine, glycine, and alanine. The fully automated assay was based on phenylisothiocyanate derivatization in the presence of internal standards followed by flow injection analysis–mass spectrometry (for glucose) and liquid chromatography–tandem mass spectrometry (for glycolytic amino acids) using a SCIEX 4000 QTrap mass spectrometer (SCIEX, Darmstadt, Germany) with electrospray ionization. Brain tissue glucose concentration was derived as the sum of hexoses detected and absolute concentration expressed as nmol/mg tissue.
To assess activities of the three rate-controlling enzymes of glycolysis (i.e., hexokinase [HK], phosphofructokinase [PFK], and pyruvate kinase [PK]), we calculated the ratios of the concentrations of the glycolytic amino acids serine, glycine, and alanine to glucose. These amino acids are biosynthetic derivatives of intermediate metabolites in glycolysis and thus their concentrations relative to glucose are an indirect measure of the net enzymatic activities catalyzing the three sequential, rate-controlling, irreversible steps of glycolysis [35]. As shown in Fig. 1, the amino acid serine is synthesized from the glycolytic intermediate 3-phosphoglycerate and can be converted to glycine after transfer of a methyl group from its side chain. The ratio of the concentrations of serine and glycine to glucose (serine 1 glycine:glucose) therefore reflects the net activities of the two irreversible enzymatic reactions in glycolysis that are upstream of 3-phosphoglycerate:
the first reaction in glycolysis, that is, the phosphorylation of glucose to glucose 6-phosphate, catalyzed by the inducible enzyme, hexokinase (abbreviated here as HK) and
the phosphorylation of fructose 6-phosphate to fructose 1, 6-bisphosphate in the presence of phosphofructokinase (abbreviated here as PFK).
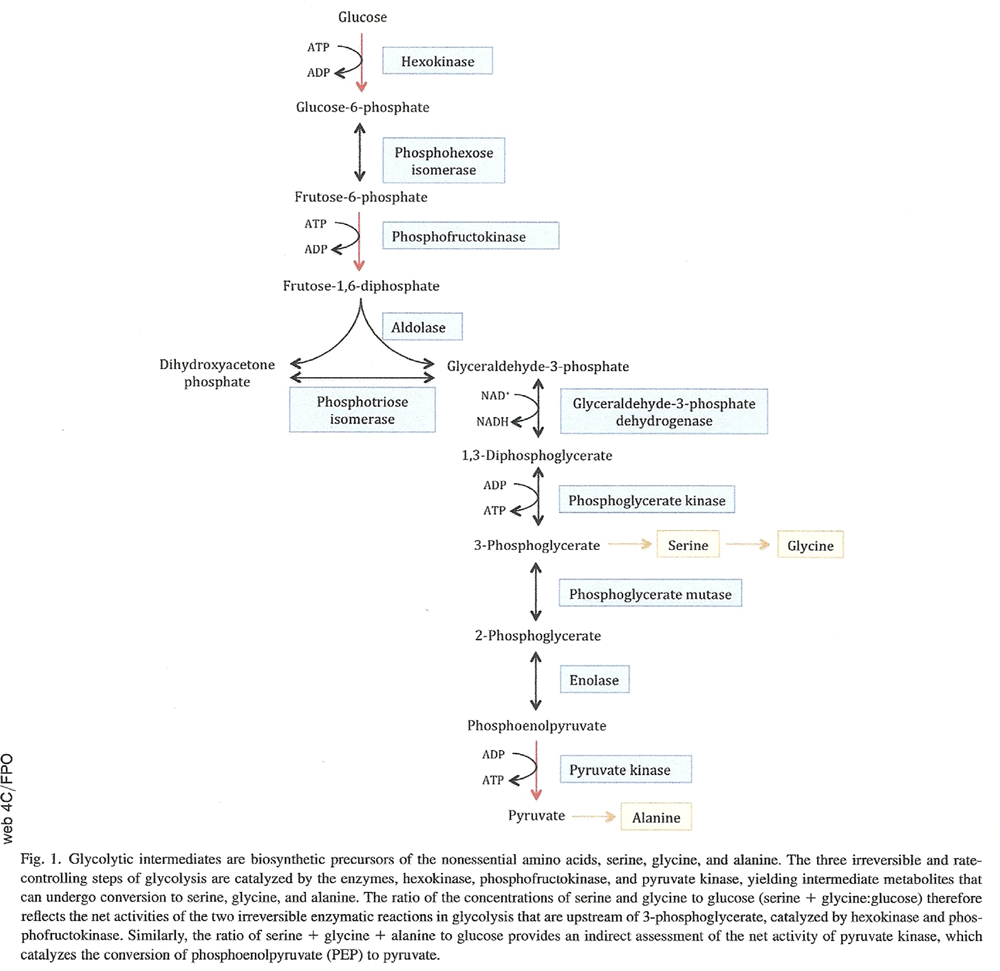
Figure 1 The conversion of phosphoenolpyruvate to pyruvate is the last step of glycolysis and is an irreversible reaction catalyzed by the enzyme pyruvate kinase (abbreviated here as PK) (Figure 1). The amino acid alanine is synthesized from pyruvate by a transamination reaction catalyzed by alanine amino transferase. Therefore, the ratio of serine 1 glycine 1 alanine to glucose provides an indirect assessment of the net activity of pyruvate kinase.
Quantification of brain tissue glucose transporter proteins
We examined protein levels of the two principal glucose transporters in the brain, that is, neuronal glucose transporter-3 (GLUT3) and glucose transporter-1 (GLUT1); the main glucose transporter in astrocytes and within vascular endothelial cells of the blood-brain barrier [36].
Label-free quantification (LFQ) of the brain proteome by LC-MS/MS was performed in the MFG using a Thermo- Fisher Scientific (San Jose, CA) Q-Exactive Plus mass spectrometer. Analysis of raw data was performed using MaxQuant 1.5.3.28 software (Max Planck Institute of Biochemistry, Martinsried, Germany) and bootstrap regressed against age and postmortem interval of each individual. In addition to protein levels of GLUT3 and GLUT1, we also quantified levels of nesprin-1, a neuronal nuclear protein. Additional details on collection of proteomic data, raw data availability, and analyses parameters are provided through the Synapse AMP-AD portal (https://www. synapse.org/#!Synapse:syn3606086) and have been reported previously [37]. Additional details on the specificity of the LFQ workflow to identify GLUT3 and GLUT1 proteins are provided in Supplementary Materials (Table-S1).
The proteomic analyses were performed on the MFG in 47 BLSA participants (N = 20 AD; N = 13 control; and N = 14 “ASYMAD”) and included 40 individuals in whom brain tissue glucose and glycolytic amino acids concentrations were also estimated by quantitative metabolomics as described in the previous section.
Statistical analyses
Differences in severity of AD pathology between groups were examined using the Mantel-Haenszel chi-square test of correlation. Proportional odds ordinal logistic models [38] are a generalization of the Wilcoxon and Kruskal-Wallis tests that allow for covariates adjustment. We used these tests to compare group differences (i.e., AD, control, and ASYMAD) in brain tissue glucose concentrations as well as ratios of glycolytic amino acids to glucose within each of the three brain regions (i.e., ITG, MFG, and cerebellum). Sensitivity analyses were conducted using sex and age at death as covariates.
A similar approach was taken for analyses of group differences in protein levels of the glucose transporters, GLUT3 and GLUT1. To confirm that observed changes in levels of the neuronal glucose transporter, GLUT3, were not driven primarily by neuronal loss, LFQ-derived protein levels of the neuronal nuclear protein, nesprin-1 [39], were used as an additional covariate in addition to sex and age at death in sensitivity analyses comparing GLUT3 protein levels between groups.
Associations between brain tissue glucose concentration, ratios of the glycolytic amino acids to glucose, and AD pathology were examined using Spearman’s rank correlation before and after adjusting for sex and age at death. Associations between protein levels of the glucose transporters and AD pathology were examined similarly with protein levels of nesprin-1 as an additional covariate in analyses involving GLUT3.
To investigate the associations between brain tissue glucose concentrations and AD pathology with longitudinal fasting plasma glucose concentrations measured during the BLSA research visits, separate linear mixed-effects models were fit with longitudinal glucose measures as the outcome and each pathology variable (i.e., CERAD and Braak score) and brain glucose concentration as the main predictors. The time of follow-up was anchored at the last measurement of fasting plasma glucose (using time of last measurement as the time origin, time = 0), with all previous longitudinal observations considered negative relative to the last measurement. This recentering of the time variable allows us to test the effects of the last plasma fasting glucose levels as well as the rates of change in fasting plasma glucose concentrations on pathology and brain tissue glucose concentration simultaneously in a single model. Other covariates included age at measurement of the last plasma fasting glucose concentration and sex. Brain tissue glucose concentration was natural log transformed and z-scored, age was mean centered and sex coded as 20.5 for female and 0.5 for male.
All the analyses were conducted in SAS 9.4 (Cary, NC).
Results
Brain tissue glucose concentration in AD

Table 1
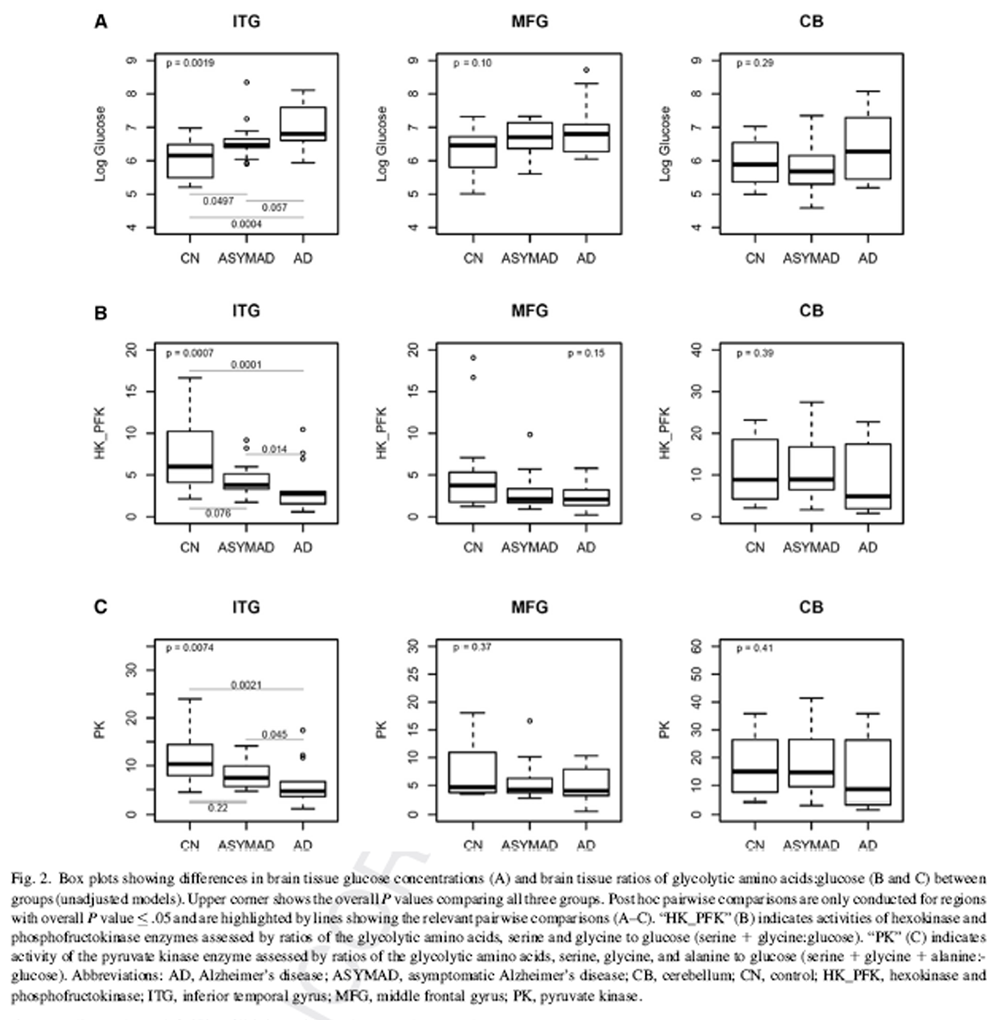
Figure 2 The demographic characteristics of BLSA participants who contributed data to the measurements of brain tissue glucose concentration are shown in Table 1. The three groups, that is, AD, controls, and “ASYMAD” did not differ significantly in age at death or the postmortem interval to autopsy. There were also no group differences in sex or APOE ε4 carrier status.
Fig. 2A summarizes results of analyses comparing brain tissue glucose concentrations between the three groups. In unadjusted models, brain tissue glucose concentrations were significantly different in the ITG (global P value for significance across groups = .0019) with tissue glucose concentration following the pattern AD > ASYMAD > control (Fig. 2A). Pairwise comparisons between the groups in the ITG showed significantly higher concentration of glucose in the AD (P = .0004) and ASYMAD groups (P = .0497) relative to controls and a trend toward higher concentration in AD relative to ASYMADs (P = .057). Results were similar after adjusting for sex and age at death. In the MFG, group differences from unadjusted models did not reach significance (global P =.10) (Fig. 2A), but after additionally adjusting for sex and age at death, results approached significance (global P value for significance across groups = .051). Pairwise comparisons between the groups in the MFG showed significantly higher concentration of glucose in the AD (P =.032) and ASYMAD groups (P =.026) relative to controls. There were no group differences in tissue glucose concentrations within the cerebellum in either unadjusted (global P value for significance across groups = .29; Fig. 2A) or adjusted models.
Brain tissue glucose concentration and AD pathology
The three groups differed significantly in the severity of both neuritic plaque and neurofibrillary tangle pathology following a linear trend (Mantel-Haenszel chi-square test of correlation between group and Braak scores: P < .0001; CERAD scores: P < .0001) with the AD group showing the highest, ASYMAD intermediate, and controls lowest levels of pathology.
Table 4 Table 4 shows correlations between brain tissue glucose concentration and measures of AD pathology. Brain tissue glucose concentration in the ITG was significantly associated with both Braak (Spearman’s r = 0.37; P = .014) and CERAD scores (Spearman’s r = 0.47; P =.0014). Tissue glucose concentrations in the MFG were significantly associated with CERAD scores (Spearman’s r = 0.39; P = .0096). No significant association was observed between glucose concentration in the cerebellum and AD pathology. These results remained similar after adjusting for sex and age at death.
Brain tissue ratios of glycolytic amino acids to glucose in AD
Fig 2B and 2C summarize results of analyses comparing brain tissue ratios of the glycolytic amino acids, serine 1 glycine to glucose; assessing hexokinase and phosphofructokinase activities (“HK_PFK”) and serine + glycine + alanine to glucose; assessing pyruvate kinase activity (“PK”) between the three groups, respectively. In unadjusted models, brain tissue HK_PFK activities were significantly different in the ITG (global P value for significance across groups = .0007) following the pattern AD < ASYMAD < control (Fig. 2B). Pairwise comparisons between the groups in the ITG showed significantly lower HK_PFK activity in the AD group relative to controls (P 5.0001) as well as lower activity in AD relative to ASYMADs (P 5.014). Similarly, in unadjusted models within the ITG, we found that brain tissue PK activity was also significantly different between the groups (ITG; global P value for significance across groups 5 .0074) with the pattern, AD , ASYMAD , control (Fig. 2C). Pairwise comparisons between the groups in the ITG showed significantly lower PK activity in the AD group relative to controls (P = .0021) as well as lower activity in AD relative to ASYMADs (P = .045). These results were similar in models additionally adjusted for sex and age at death. In the MFG, group difference in brain tissue HK_PFK activities did not reach statistical significance in the unadjusted model (Fig. 2B) but showed a trend toward significance in adjusted models (HK_PFK; global P value for significance across groups = .058). There were no significant group differences in PK activity in the MFG in either unadjusted (PK; global P value for significance across groups = .368; Fig. 2C) or adjusted models. There were no group differences in brain tissue activities of either HK_PFK or PK within the cerebellum in either unadjusted (HK_PFK; global P value for significance across groups = .394; PK; global P value for significance across groups - .410, Fig 2B and 2C) or adjusted models.
Brain tissue ratios of glycolytic amino acid to glucose and AD pathology
Table 4 shows correlations between ratios of the glycolytic amino acids, serine + glycine to glucose; assessing hexokinase and phosphofructokinase activities (i.e., “HK_PFK”) and serine + glycine + alanine to glucose; assessing pyruvate kinase activity (i.e., “PK”) against measures of AD pathology. Brain tissue HK_PFK activities in the ITG were significantly associated with both Braak (Spearman’s p = –0.37; P = .016) and CERAD (Spearman’s p = –0.52; P = .0004) scores. Similarly, PK activity in the ITG was also significantly associated with both Braak (Spearman’s p = –0.46; P = .0093) and CERAD (Spearman’s p = –0.55; P = .0010) scores. In the MFG, HK_PFK activities were significantly associated with the CERAD (Spearman’s p = –0.38; P = .013) score. These results remained similar after adjusting for sex and age at death.
Brain tissue protein levels of glucose transporters (GLUT3 and GLUT1)
Table 2
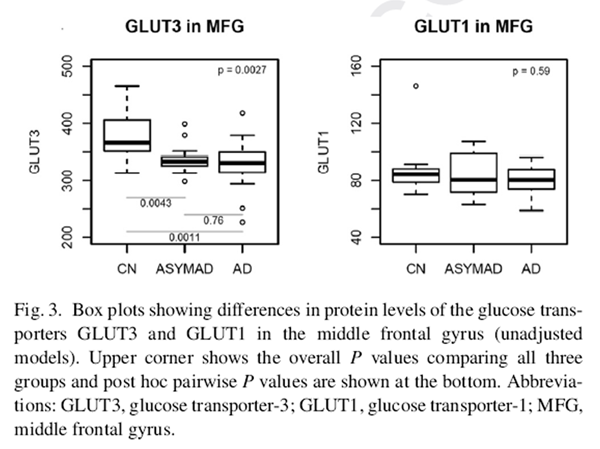
Figure 3 The demographic characteristics of BLSA participants who contributed data to the proteomic analyses of brain tissue glucose transporter levels are shown in Table 2. The three groups, that is, AD, controls, and “ASYMAD,” did not differ significantly in age at death or the postmortem interval to autopsy. There were also no group differences in sex or APOE ε4 carrier status.
Fig. 3 summarizes results of analyses comparing brain tissue glucose transporter protein levels between the three groups in the MFG. In unadjusted models, brain tissue protein levels of the neuronal glucose transporter GLUT3 were significantly different across the groups (global P value for significance = .0027) (Fig. 3). Pairwise comparisons showed significantly lower protein levels of GLUT3 in the AD (P = .0011) and ASYMAD (P = .0043) groups relative to controls. These results remained similar in models adjusted for sex, age at death, and levels of the neuronal nuclear protein, nesprin-1. No significant differences were observed in protein levels of the astrocytic glucose transporter GLUT1.
Brain tissue protein levels of glucose transporters and AD pathology
Protein levels of GLUT3 were significantly associated with both Braak (Spearman’s p = –0.40; P = .0051) and CERAD (Spearman’s p = –0.42; P = .0033) scores in the MFG. There were no significant associations between protein levels of GLUT1 and Braak (Spearman’s p = 0.038; P = .80) or CERAD (Spearman’s p = –0.21; P - .16) scores. These results remained unchanged in models adjusted for sex, age at death, and protein levels of the neuronal nuclear protein, nesprin-1 (Table 4).
Associations between plasma fasting glucose and brain tissue glucose concentrations
Table 3
Table 5 Table 3 summarizes the plasma fasting glucose measurements that were used in the analyses examining their relationship with brain tissue glucose concentrations. A total of 445 plasma fasting glucose measures were available in the 43 BLSA participants who also contributed measures of brain tissue glucose concentration described previously. The average follow-up interval for the plasma glucose measurements was 19.1 years, and the average interval between the last available fasting plasma glucose assessment and death was 5 years.
In linear mixed models adjusted for age at the last available plasma glucose measurement and sex, we found that both the last plasma fasting glucose concentration and longitudinal trajectories of change in plasma fasting glucose were associated with brain tissue glucose concentrations in all three regions examined, that is, ITG, MFG, and cerebellum (Table 5). The direction of this association indicates that higher levels (i.e., last measured fasting plasma glucose concentration) as well as greater increases over time in plasma fasting glucose are associated with higher brain tissue glucose concentrations. These results were similar in models that were further adjusted for clinical status, that is, AD/control/ ASYMAD.
In sensitivity analyses, we excluded 26 plasma fasting glucose values in the diabetic range, that is, ≥126 mg/dL; longitudinal associations remained unchanged in the MFG and ITG; however, cross-sectional associations only remained significant in the ITG. When we examined the relationship between plasma fasting glucose concentrations and AD pathology, there were no significant associations with either Braak or CERAD scores.
Discussion
In this study, we have demonstrated that abnormalities in brain glucose homeostasis are intrinsic to AD pathogenesis and may begin several years before the onset of clinical symptoms. We first showed that brain regions vulnerable to amyloid deposition and neurofibrillary pathology show significantly higher tissue glucose concentrations in AD. Moreover, higher concentrations of brain tissue glucose are associated with greater severity of both amyloid plaque deposition and neurofibrillary pathology.
To further investigate abnormalities in glucose metabolism associated with higher glucose concentrations in brain regions vulnerable to AD pathology, we used concentrations of the glycolytic amino acids, serine, glycine, and alanine to assess the three rate-controlling, irreversible steps of glycolysis. Besides the generation of energy through ATP from the breakdown of glucose, intermediates of glycolysis serve as biosynthetic precursors of several metabolites including the nonessential amino acids, serine, glycine, and alanine (Fig. 1) [35]. The ratios of these glycolytic amino acids to that of glucose, the primary substrate of glycolysis, are hence useful as indirect measures of the enzymatic activities of hexokinase, phosphofructokinase, and pyruvate kinase. Our results indicate that the activities of these principal rate-controlling enzymes of glycolysis are significantly reduced in the ITG and MFG, but not cerebellum, of individuals with AD. Moreover, lower activities of these enzymes are associated with greater severity of both neurofibrillary and amyloid pathology.
Next, using mass spectrometry–based proteomics in brain tissue samples from the same participants, we showed that protein levels of the neuronal glucose transporter, GLUT3, are significantly reduced in AD and that lower levels of GLUT3 protein reflect greater severity of both amyloid plaque and neurofibrillary tangle pathology. Finally, we demonstrated that longitudinal increases in fasting plasma glucose levels, measured up to four decades before death, are associated with higher brain tissue glucose concentrations globally.
To the best of our knowledge, this report is the first to measure brain tissue glucose concentrations and assess glycolytic flux to demonstrate their relationships with both severity of AD pathology and the expression of AD symptoms. Including brain tissue samples from “ASYMAD” individuals who represent an intermediate group in the gradation of neuropathology from controls to AD patients in the absence of cognitive impairment during life allowed us to relate measures of brain glucose concentration and glycolytic flux to incremental levels of AD pathology and symptom expression. Equally importantly, by measuring glucose concentrations in brain regions both vulnerable to distinct pathological features of AD, that is, MFG (amyloid deposition) and ITG (tau accumulation) as well as in a region relatively resistant to AD pathology, that is, cerebellum [34], we were able to determine whether the observed alterations in brain glucose concentrations and abnormalities in glycolysis were related to ADdefining pathological processes.
Our observation that in the ITG, brain glucose concentrations are highest in AD, lowest in controls, and intermediate in ASYMADs suggests that there may be regionally specific threshold levels of tissue glucose beyond which the accumulation of AD pathology triggers the expression of clinical symptoms. Complementing these observations on brain glucose concentrations in the ITG, we also observed that activities of the three rate-controlling enzymes of glycolysis, that is, hexokinase, phosphofructokinase, and pyruvate kinase, are lowest in AD, highest in controls, and intermediate in ASYMADs, suggesting that regionally specific abnormalities in glycolytic flux may lead to the build-up of brain glucose levels, evolution of AD pathology, and ultimately the development of AD symptoms. Furthermore, the absence of significant group differences in either brain tissue glucose concentration or glycolytic flux in the cerebellum, a region that is relatively spared of classical AD pathology, suggests that these abnormalities are specifically linked to the defining pathological hallmarks of AD. This interpretation is further strengthened by the significant associations between concentrations of brain tissue glucose and measures of glycolytic flux with severity of AD pathology in both the ITG and the MFG.
Our proteomic analysis of brain tissue from the same groups of participants in the BLSA autopsy sample allowed us to further explore potential mechanisms underlying alterations in brain glucose concentration in AD.We found that protein levels of the neuronal glucose transporter, GLUT3, are significantly lower in AD and ASYMAD brains relative to controls. Furthermore, lower protein levels of GLUT3 are associated with greater severity of both neuritic plaque and neurofibrillary tangle pathology. Equally importantly, we find that both the group differences in GLUT3 protein levels as well as their association with AD pathology are independent of neuronal loss as they remain significant even after adjustment for levels of the neuronal nuclear protein nesprin-1 [39]. This suggests that lower GLUT3 protein levels in AD are likely to reflect early changes in the evolution of AD pathophysiology, rather than downstream consequences of neurodegeneration. Together with the observation that protein levels of the astrocytic glucose transporter GLUT1 are unchanged, our results implicate a specific loss of the neuronal glucose transporter GLUT3 as an integral feature of AD pathogenesis.
Our findings also complement and extend previous functional neuroimaging studies using 18FDG-PET showing that decreases in neuronal glucose uptake may be an early feature of AD pathogenesis [19–22, 40]. These studies show a characteristic pattern of hypometabolism affecting primarily the parietotemporal, posterior cingulate, and frontal cortices with relative sparing of the cerebellum, thalamus, and basal ganglia [22]. This pattern of lower cerebral glucose uptake on FDG-PET imaging therefore appears consistent with our current observation of higher brain tissue glucose levels in the frontal and temporal cortices in AD but sparing the cerebellum. Previous studies have shown that the density of the neuronal GLUT3 transporter in the brain is coupled to local cerebral glucose utilization [41]. Our current results indicate that lower cerebral glucose utilization, as reflected in lower rates of glycolysis and higher brain tissue glucose concentrations, may thus downregulate GLUT3 protein levels, especially in brain regions vulnerable to AD pathology.
Previous studies of GLUT3 and GLUT1 protein levels in the AD brain have relied on small sample sizes and important methodological differences with our current report merit consideration. Simpson and colleagues measured GLUT3 and GLUT1 protein levels using immunoblotting and autoradiography from a sample of 12 AD and 12 control brains [42]. The mean age of the control group (56 ± 22 years) in their report was considerably lower than that of the AD group (76 ± 5 years). Although they observed overall reductions in GLUT1 and GLUT3 protein levels averaged across several regions, considerable interindividual variability made comparisons of regional differences in glucose transporter levels difficult. Using cytochalasin B binding assays, Kalaria and colleagues demonstrated lower glucose transporter levels in AD brains compared to controls [43]. However, as cytochalasin B is a nonspecific blocker of both GLUT1 and GLUT3 transporters [44], a reliable assessment of differences in neuronal versus astrocytic glucose transporter levels was not feasible. Moreover, in both these studies, the absence of “high-pathology controls” or “ASYMADs” as in our current report precludes assessment of changes in glucose transporter levels in relation to severity of AD pathology and expression of AD symptoms. A definitive assessment of regional differences in GLUT3 and GLUT1 levels in AD may require a larger sample size and proteomic profiling across several brain regions.
It is important to note that our measure of brain tissue glucose is based on mass spectrometric quantification of total hexose levels transported from the circulation. Glucose is the predominant hexose in blood and is present in far greater concentrations (5450.0 ± 1200 µM) than other hexoses such as fructose (46.26 ± 25.22 µM), galactose (88.3 ± 34.7 µM), and mannose (64.0 ± 12.0 µM) [45–47]. Therefore, the total concentration of brain hexoses measured in our samples is likely to comprise predominantly of glucose.
Given previous epidemiological evidence that insulin resistance and diabetes are associated with increased risk of AD [1–3], we also examined whether peripheral glucose levels measured decades before death may influence brain tissue glucose concentration. We have previously shown that this relationship does not appear to be mediated by a direct effect of blood glucose concentrations in either brain amyloid deposition or neurofibrillary pathology [48]. In the current report, we found that higher concentrations of plasma fasting glucose measured before death as well as greater increases in fasting plasma glucose over time were associated with higher brain tissue glucose concentrations in all three regions examined. In additional sensitivity analyses, we further confirmed that the longitudinal associations between fasting plasma glucose and brain tissue glucose concentrations are not driven by individuals with diabetes. These findings open up new lines of investigation in appropriate animal models of AD where longitudinal analyses of brain tissue and blood glucose concentrations in relation to accumulating AD pathology may provide important insights into the role of glucose dysregulation in early stages of AD pathogenesis. Such experimental studies are essential to test both causal relationships confirm the temporal sequence of molecular events related to glucose dysregulation and AD pathogenesis in humans. It would also be important to test whether abnormalities of brain glucose utilization are a specific feature of AD pathogenesis or may be shared by other neurodegenerative diseases. The lack of brain and blood tissue samples from well-characterized cohorts representing other non-AD neurological diseases is a limitation of our present report.
Considering our results together, we propose that failure of neuronal glucose utilization due to impaired glycolysis is a fundamental feature of AD. At regionally specific threshold levels of brain glucose and impaired glycolytic flux, accumulating pathology in vulnerable brain regions triggers the onset of AD symptoms. Our results have important translational implications and set the stage for future studies that may uncover therapeutic interventions targeting brain glucose dysregulation in AD.
Acknowledgments
The authors are grateful to participants in the Baltimore Longitudinal Study of Aging for their invaluable contribution. This research was supported in part by the Intramural Research Program of the NIH, National Institute on Aging. Support was provided by the Accelerating Medicine Partnership AD grant (grant number: U01AG046161-02), the NINDS Emory Neuroscience Core (grant number: P30NS055077), and the Emory Alzheimer’s Disease Research Center (grant number: P50AG025688). The Intramural Research Program of the National Institute on Aging (NIH) had no role in study design; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
RESEARCH IN CONTEXT
Systematic review: We reviewed (using PubMed) all publications describing abnormalities of glucose metabolism in Alzheimer’s disease (AD). It is unclear whether glucose dysregulation in the brain is related to severity of AD pathology and symptom expression. Similarly, the relationships between longitudinal changes in blood glucose concentration and brain tissue glucose levels are unclear. We measured brain tissue concentrations of glucose and the glycolytic amino acids, serine, glycine, and alanine as well as protein levels of the neuronal (GLUT3) and astrocytic (GLUT1) glucose transporters in the autopsy sample of the Baltimore Longitudinal Study of Aging. Higher brain tissue glucose concentration, reduced glycolytic flux, and lower levels of GLUT3 are related to severity of AD pathology and the expression of AD symptoms. Increasing fasting plasma glucose levels are associated with higher brain tissue glucose concentrations.
Interpretation: Abnormalities in brain glucose homeostasis are intrinsic to AD pathogenesis and begin several years before clinical symptoms.
Future directions: These results set the stage for future studies testing brain glucose dysregulation as a target of disease-modifying interventions in AD.
References:
Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O’Brien PC, et al.
Risk of dementia among persons with diabetes mellitus: a population-based cohort study.
Am J Epidemiol 1997;145:301–8.Luchsinger JA, Tang MX, Stern Y, Shea S, Mayeux R.
Diabetes mellitus and risk of Alzheimer’s disease and dementia with stroke in a multiethnic cohort.
Am J Epidemiol 2001;154:635–41.Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM.
Diabetes mellitus and the risk of dementia: The Rotterdam Study.
Neurology 1999;53:1937–42.Chen Z, Zhong C.
Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies.
Prog Neurobiol 2013;108:21–43.Hoyer S.
Brain glucose and energy metabolism abnormalities in sporadic Alzheimer disease. Causes and consequences: an update.
Exp Gerontol 2000;35:1363–72.Ahmed S, Mahmood Z, Zahid S.
Linking insulin with Alzheimer’s disease: emergence as type III diabetes.
Neurol Sci 2015;36:1763–9.Chami B, Steel AJ, De La Monte SM, Sutherland GT.
The rise and fall of insulin signaling in Alzheimer’s disease.
Metab Brain Dis 2016; 31:497–515.Freude S, Schilbach K, Schubert M.
The role of IGF-1 receptor and insulin receptor signaling for the pathogenesis of Alzheimer’s disease: from model organisms to human disease.
Curr Alzheimer Res 2009; 6:213–23.Hokama M, Oka S, Leon J, Ninomiya T, Honda H, Sasaki K, et al.
Altered expression of diabetes-related genes in Alzheimer’s disease brains: the Hisayama study.
Cereb Cortex 2014;24:2476–88.Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, et al.
Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline.
J Clin Invest 2012;122:1316–38.de la Monte SM.
Type 3 diabetes is sporadic Alzheimers disease: minireview.
Eur Neuropsychopharmacol 2014;24:1954–60.de la Monte SM, Wands JR.
Alzheimer’s disease is type 3 diabetesevidence reviewed.
J Diabetes Sci Technol 2008;2:1101–13.Claxton A, Baker LD, Hanson A, Trittschuh EH, Cholerton B, Morgan A, et al.
Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or earlystage Alzheimer’s disease dementia.
J Alzheimers Dis 2015; 44:897–906.Claxton A, Baker LD, Wilkinson CW, Trittschuh EH, Chapman D, Watson GS, et al.
Sex and ApoE genotype differences in treatment response to two doses of intranasal insulin in adults with mild cognitive impairment or Alzheimer’s disease.
J Alzheimers Dis 2013; 35:789–97.Reger MA, Watson GS, Green PS, Wilkinson CW, Baker LD, Cholerton B, et al.
Intranasal insulin improves cognition and modulates beta-amyloid in early AD.
Neurology 2008;70:440–8.Benarroch EE.
Brain glucose transporters: implications for neurologic disease.
Neurology 2014;82:1374–9.Simpson IA, Carruthers A, Vannucci SJ.
Supply and demand in cerebral energy metabolism: the role of nutrient transporters.
J Cereb Blood Flow Metab 2007;27:1766–91.Joost HG, Thorens B.
The extended GLUT-family of sugar/polyol transport facilitators: nomenclature, sequence characteristics, and potential function of its novel members (review).
Mol Membr Biol 2001; 18:247–56.Herholz K.
Cerebral glucose metabolism in preclinical and prodromal Alzheimer’s disease.
Expert Rev Neurother 2010;10:1667–73.Hunt A, Schonknecht P, Henze M, Seidl U, Haberkorn U, Schroder J.
Reduced cerebral glucose metabolism in patients at risk for Alzheimer’s disease.
Psychiatry Res 2007;155:147–54.Jagust WJ, Haan MN, Eberling JL,Wolfe N, Reed BR.
Functional imaging predicts cognitive decline in Alzheimer’s disease.
J Neuroimaging 1996;6:156–60.Mosconi L.
Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD.
Eur J Nucl Med Mol Imaging 2005;32:486–510.Ferrucci L.
The Baltimore Longitudinal Study of Aging (BLSA): a 50- year-long journey and plans for the future.
J Gerontol A Biol Sci Med Sci 2008;63:1416–9.Shock NW, Gruelich R, Andres R, Arenberg D, Costa PT, Lakatta E, et al.
Normal human aging. The Baltimore Longitudinal Study of Aging.
Washington, DC: US Government Printing Office; 1984.O’Brien RJ, Resnick SM, Zonderman AB, Ferrucci L, Crain BJ, Pletnikova O, et al.
Neuropathologic studies of the Baltimore Longitudinal Study of Aging (BLSA).
J Alzheimers Dis 2009;18:665–75.Gamaldo A, Moghekar A, Kilada S, Resnick SM, Zonderman AB, O’Brien R.
Effect of a clinical stroke on the risk of dementia in a prospective cohort.
Neurology 2006;67:1363–9.Diagnostic and Statistical Manual of Mental Disorders, DSM-III-R.
Washington, D.C.: American Psychiatric Association. Q6McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM.
Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDAWork Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease.
Neurology 1984;34:939–44.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al.
he Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease.
Neurology 1991;41:479–86.Braak H, Braak E.
Neuropathological stageing of Alzheimer-related changes.
Acta neuropathol 1991;82:239–59.Troncoso JC, Zonderman AB, Resnick SM, Crain B, Pletnikova O, O’Brien RJ.
Effect of infarcts on dementia in the Baltimore longitudinal study of aging.
Ann Neurol 2008;64:168–76.Iacono D, Resnick SM, O’Brien R, Zonderman AB, An Y, Pletnikova O, et al.
Mild cognitive impairment and asymptomatic Alzheimer disease subjects: equivalent beta-amyloid and tau loads with divergent cognitive outcomes.
J Neuropathol Exp Neurol 2014;73:295–304.Metter EJ, Windham BG, Maggio M, Simonsick EM, Ling SM, Egan JM, et al.
Glucose and insulin measurements fromthe oral glucose tolerance test and mortality prediction.
Diabetes care 2008;31:1026–30.Larner AJ.
The cerebellum in Alzheimer’s disease.
Dement Geriatr Cogn Disord 1997;8:203–9.Berg JM, Tymoczko JL, Stryer L, Stryer L.
Biochemistry. 5th ed.
New York: W.H. Freeman; 2002.Vannucci SJ, Maher F, Simpson IA.
Glucose transporter proteins in brain: delivery of glucose to neurons and glia.
Glia 1997;21:2–21.SeyfriedNT,DammerEB, SwarupV,NandakumarD,DuongDM,YinL, et al.
A Multi-network Approach Identifies Protein-Specific Co-expression in Asymptomatic and Symptomatic Alzheimer’s Disease.
Cell Syst 2017;4:60–72.e4.Harrell FE.
Regression Modeling Strategies with Applications to Linear Models, Logistic and Ordinal Regression, and Survival Analysis.
Cham: Springer; 2015.Dammer EB, Duong DM, Diner I, Gearing M, Feng Y, Lah JJ, et al.
Neuron enriched nuclear proteome isolated from human brain.
J Proteome Res 2013;12:3193–206.Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, et al.
Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia.
Proc Natl Acad Sci U S A 2004; 101:284–9.Duelli R, Kuschinsky W.
Brain glucose transporters: relationship to local energy demand.
News Physiol Sci 2001;16:71–6.Simpson IA, Chundu KR, Davies-Hill T, Honer WG, Davies P.
Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease.
Ann Neurol 1994; 35:546–51.Kalaria RN, Harik SI.
Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in Alzheimer disease.
J Neurochem 1989;53:1083–8.Dick AP, Harik SI, Klip A, Walker DM.
Identification and characterization of the glucose transporter of the blood-brain barrier by cytochalasin B binding and immunological reactivity.
Proc Natl Acad Sci U S A 1984;81:7233–7.Wahjudi PN, Patterson ME, Lim S, Yee JK, Mao CS, Lee WN.
Measurement of glucose and fructose in clinical samples using gas chromatography/mass spectrometry.
Clin Biochem 2010;43:198–207.Lentner C.
Geigy scientific tables, Vol. 1.
Basle: Ciba-Geigy; 1981.Wu AHB.
Tietz clinical guide to laboratory tests.
St. Louis: Saunders Elsevier; 2006.Thambisetty M, Jeffrey Metter E, Yang A, Dolan H, Marano C, Zonderman AB, et al.
Glucose intolerance, insulin resistance, and pathological features of Alzheimer disease in the Baltimore Longitudinal Study of Aging.
JAMA Neurol 2013;70:1167–72.
Return to NUTRITION
Return to ALZHEIMER's
Since 2-12-2018
| Home Page | Visit Our Sponsors | Become a Sponsor |
Please read our DISCLAIMER |