

Misfolded Proteins, Endoplasmic Reticulum Stress and Neurodegeneration This section is compiled by Frank M. Painter, D.C.
Send all comments or additions to: Frankp@chiro.org


FROM: Curr Opin Cell Biol. 2004 (Dec); 16 (6): 653–662 ~ FULL TEXT
Veena Theendakara & Clare A. Peters-Libeu & Dale E. Bredesen
The Buck Institute for Research on Aging,
8001 Redwood Blvd,
Novato, CA, 94945, USA.
rrao@buckinstitute.orgThe accumulation of misfolded proteins (e.g. mutant or damaged proteins) triggers cellular stress responses that protect cells against the toxic buildup of such proteins. However, prolonged stress due to the buildup of these toxic proteins induces specific death pathways. Dissecting these pathways should be valuable in understanding the pathogenesis of, and ultimately in designing therapy for, neurodegenerative diseases that feature misfolded proteins.
From the FULL TEXT Article:
Introduction
Neurodegenerative disorders such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS) and prion protein diseases all share a common feature: the accumulation and aggregation of misfolded proteins [1–3]. The presence of misfolded proteins elicits cellular responses that include an endoplasmic reticulum (ER) stress response that may protect cells against the toxic buildup of misfolded proteins [3–7]. Accumulation of these proteins in excessive amounts, however, overwhelms the ‘cellular quality control’ system and impairs the protective mechanisms designed to promote correct folding and degrade faulty proteins, ultimately leading to organelle dysfunction and cell death [1–10].
Since the degradation of cellular proteins in general is coupled, via the ubiquitin-mediated proteasomal degradation pathway, to ER dislocation of many ER substrates [3, 9, 10], any conditions that block ER retrotranslocation of proteins and/or proteasome function and degradation may also result in the accumulation of misfolded protein substrates within the ER. Thus, misfolded proteins trigger the ER stress response, whether the misfolded proteins build up within, or outside, the ER (e.g. in the nucleus or cytosol) and transmit toxic responses across cellular compartments. Misfolded proteins may be deposited as microscopically visible inclusion bodies or plaques within cells or in extracellular spaces, and have a high propensity to interact with a wide range of cellular targets to elicit cellular toxicity [1]. Toxicity may arise due to one or more of a number of factors: inhibition of synaptic function; loss of synpases leading to disruption of neuronal functions; sequestration of critical cellular chaperones and vital transcription factors by misfolded proteins; interference with numerous signal-transduction pathways; alteration of calcium homeostasis; release of free radicals and consequent oxidative damage; dysfunction of the protein degradation pathway through the ubiquitinproteasome system; and/or induction of cell-death proteases leading to programmed cell death.
This review highlights recent advances in understanding the complex regulation of ER stress and the unfolded protein response (UPR), and their relevance to apoptosis and neurodegenerative diseases that feature misfolded proteins.
Endoplasmic reticulum stress
The ER is a principal site for biosynthesis of proteins, steroids, cholesterol and other lipids. It also serves as a site of calcium storage and calcium signaling. The ER serves several important functions, including the post-translational modification, folding and assembly of newly synthesized secretory proteins, and its proper functioning is essential to cell survival. Nascent secretory and membrane proteins that are translocated into the ER lumen start to fold co-translationally. Post-translational modification, including proper folding, assembly of individual subunits and oligomerization, is necessary for optimal function. Each co-translational and post-translational step requires specific and sequential interaction with a distinct chaperone protein. These chaperone proteins perform diverse roles, including catalyzing isomerization reactions, maintaining proteins in a folding-competent state, preventing luminal protein transit through the secretory pathway, and regulating retro-translocation of misfolded proteins for degradation. Besides providing a unique oxidizing environment for protein folding, the ER also plays a critical role in discriminating between normal (native) and abnormal (mutant) proteins. As a membranous compartment associated with the critical functions mentioned above, the ER is extremely sensitive to changes that affect its structure, integrity and function. [6, 8, 11, 12].
Thus, changes in calcium homeostasis leading to calcium depletion from the ER lumen, inhibitors of protein glycosylation, inhibitors of disulfide-bond formation, virus infection, hypoxia, ischemia and growth factor depletion can all disrupt protein synthesis, translation and folding, resulting in unfolded or misfolded proteins. The accumulation of unfolded and/or misfolded proteins causes an imbalance between the synthesis of new proteins and the ER’s ability to process newly synthesized proteins, resulting in the failure of the ER to cope with the excess protein load, which is termed ‘ER stress’ [5, 8, 13–15]. Cells in turn activate an integrated intracellular signaling cascade — the ‘unfolded protein response’ — to avert ER stress.
The unfolded-protein response
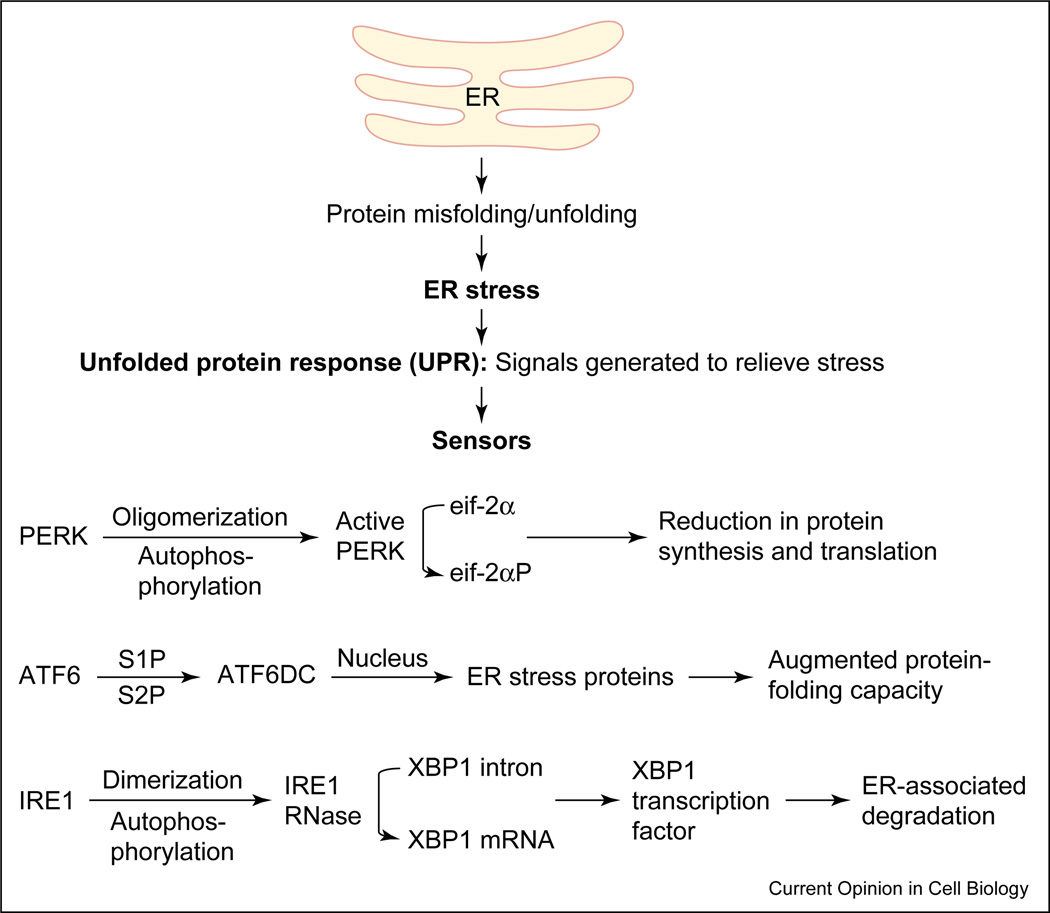
Figure 1 In cells from all organisms, ER stress may elicit a set of cellular responses collectively referred to as the UPR [11, 12, 16, 17] (Figure 1). UPR activation results in, first, a transient attenuation in the rate of protein synthesis, second, an upregulation of genes encoding chaperones and other proteins that prevent polypeptide aggregation and participate in polypeptide folding, and third, retro-translocation and degradation of ER-localized proteins. These cellular responses minimize the accumulation and aggregation of misfolded proteins by increasing the capacity of the ER machinery for folding and degradation [11, 16, 18] (Figure 1).
UPR and the molecules involved in the UPR signaling pathway were first described in the yeast Saccharomyces cerevisiae. In eukaryotes, the UPR is a is a coordinated, regulated response involving three sensor proteins: PERK (PKR-like ER kinase), IRE1 (inositol-requiring transmembrane kinase/endoribonuclease) and ATF6 (activating transcription factor 6) [14] (Figure 1). The presence of misfolded proteins and UPR activation triggers PERK oligomerization and autophosphorylation. Active PERK phosphorylates eukaryotic translation initiation factor 2α (eIF2α), rendering it inactive and blocking protein translation. Inactivation of eIF2α prevents the further influx of nascent proteins into the ER lumen, thus limiting the incoming protein load [19–22] (Figure 1).
The accumulation of misfolded proteins and UPR activation also lead to the translocation of ATF6 to the Golgi compartment, where it undergoes regulated intramembrane proteolysis (RIP) by proteases S1P and S2P, yielding a free cytosolic domain that, following nuclear translocation, triggers transcriptional upregulation of ER chaperone proteins like GRP78 and of bZIP transcription factors like CHOP/GADD153 [23, 24, 25, 26, 27, 28–30]. The chaperone proteins facilitate and promote the productive folding of proteins and protein complexes, maintaining them in a folding-competent state and preventing their aggregation. UPR activation also induces homodimerization, autophosphorylation and activation of IRE1, an ER resident transmembrane serine/ threonine kinase receptor protein that also possesses an intrinsic endoribonuclease activity. Activated IRE1 cleaves a preformed substrate mRNA at two sites through its endoribonuclease action, resulting in the removal of a 26-nucleotide intron from a target mRNA [31–33]. The two ends of the cleaved mRNA are ligated together by tRNA ligase and the newly formed mRNA encodes a transcription factor X-box-binding protein (XBP-1). XBP-1 binds and activates the promoters of several ER-stress-inducible target genes that facilitate the retro-translocation and ER-associated degradation of misfolded proteins [26, 27, 34]. Thus, all of the above-mentioned specific signaling pathways that constitute the UPR operate to ensure that the protein folding capacity of the ER is not overwhelmed (Figure 1).
GRP78: the master regulator
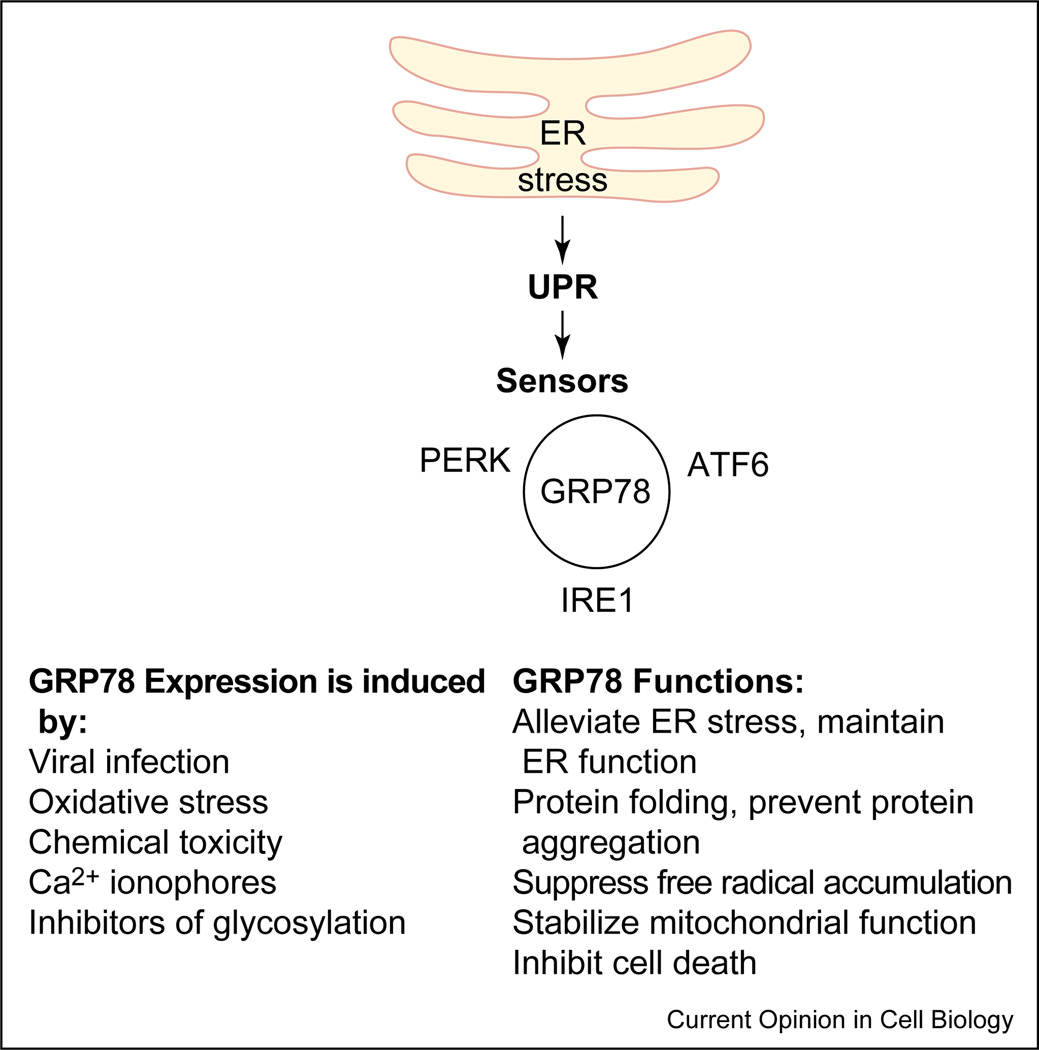
Figure 2 The glucose-regulated proteins (GRPs) are Ca2+-binding chaperone proteins with protective properties whose transcription is induced in response to several stimuli that disrupt ER structure and function [6, 17]. One of the best-characterized glucose-regulated proteins is GRP78, a 78 kD protein also referred to as BiP. The induction of GRP78 is required to block ER stress signals, maintain ER function and integrity, ensure protein folding and protect cells from misfolded protein toxicity [6, 17]. It has been well established that diverse misfolded proteins that accumulate in the cell trigger the same ‘quality control’ pathway, mainly by converging on GRP78, the ‘master molecule’. GRP78 binds to all three ER stress sensors (PERK, IRE1 and ATF6) through its peptide-binding domains, and keeps them in an inactive conformation [18, 30, 35, 36] (Figure 2). The peptide-binding domain of GRP78 also serves as the binding region for misfolded proteins. When misfolded proteins accumulate in the cell, they bind to GRP78 and disrupt its interaction with these proximal stress sensors [11, 16, 18, 36]. While free IRE1 and PERK homodimerize and undergo autophosphorylation and activation, ATF6 transits to the Golgi for proteolytic activation.
Although GRP78 exists as an ER lumen protein [6, 17], several reports have suggested that GRP78 and its mutant forms may alternatively be expressed on the cell surface, exist as ER transmembrane proteins, or redistribute to the cytosol and nucleus [37–41, 42, 43]. Recent studies indicate that either a cytosolic pool of GRP78, or a subpopulation of GRP78 existing as an ER transmembrane protein, may associate with caspases-7 and -12 and prevent their activation and release [42, 43]. These studies highlight the importance of GRP78 as an anti-apoptotic protein, and provide a link between ER stress, the unfolded protein response and the cell death program
] [77, 78, 81]. A recent study also implicates human caspase-4 in ER-stress-induced cell death. Caspase-4 is primarily activated in ER-stress-induced apoptosis and may function as an ER-stress-specific caspase in humans [82].
ER-stress-induced cell death
As stated earlier, prolonged ER stress and UPR activation completely overwhelm the cellular protective mechanisms, ultimately triggering cell death. Although there are several reports implicating various molecules as mediators of ER-stress-induced cell death, the pathway that links ER stress to programmed cell death is still relatively poorly understood. It has generally been assumed that the elimination of cells undergoing severe ER stress is desirable at the organismal level, and the upregulation of GRP78 in some malignant tumors is compatible with (although not proof of) this notion [43, 44] In any case, prolonged ER stress is indeed coupled to specific independent death pathways, as well as triggering cross-talk between the intrinsic and extrinsic apoptotic pathways.
Studies from multiple laboratories have uncovered the roles of several ER-stress-induced cell death modulators and effectors through the use of biochemical, pharmacological and genetic tools. The ER-stress-induced cell death modulators include (but are not limited to) members of the Bcl-2 family (Bcl-2, Bcl-xL, Bax, Bad, Bik, Noxa, and Bak) [45–47], p29Bap31 [48–50], c-Abl tyrosine kinase [51], and p53-dependent gene products like PUMA/Bbc3 and scotin [52, 53]. Bcl-2 family members including Bcl-2, Bcl-xL, Bax, Bad, Bik and Bak have been shown to be associated with the ER, suggesting the involvement of the Bcl-2 family proteins in ER calcium homeostasis and ER-stress-induced cell death [54, 55, 56–61].
CHOP/Gadd153, a transcription factor induced during ER stress and subsequently activated by p38 MAP (mitogen-associated protein) kinase, may also function as an ER-stress-induced cell death modulator [6, 13, 62, 63]. Deletion of the CHOP gene leads to an attenuation in the cell death usually induced by ER stress [64, 65]. Although the identification of the downstream target genes that respond to CHOP/Gadd153 is still unclear, it has been suggested that Gadd153 may promote ER-stress-induced cell death by down-regulating Bcl-2 expression [65].
Another ER-stress-induced effector protein is BAP31, an ER transmembrane protein that is cleaved by caspase-8, leading to ER calcium release and subsequent uptake by mitochondria, followed by mitochondrial cytochrome c release, further caspase activation and apoptosis. Thus, Bap31 appears be a coordinator of cell death signals between the extrinsic pathway, the ER and the mitochondrial pathway(s) [45, 66, 67].
Recent data have also implicated the calcium-binding protein apoptosis-linked gene 2 (ALG-2) and valosin-containing protein (VCP) as mediators of ER-stress-induced programmed cell death (PCD). However, there is a growing realization that some such mediators may be specific for a given inducer or set of inducers: for example, ALG-2 was found to mediate ER-stress-induced apoptosis when thapsigargin, but not tunicamyin or brefeldin-A, was the inducer; VCP, however, was found to be a mediator for all three inducers [68]. Together with caspase-12, caspase-9, caspase-7, ATP and Ca2+ (and, potentially, other molecules), VCP and ALG-2 trigger ER-stress-induced cell death [68]. VCP also functions as a sensor of abnormally folded proteins, and has been shown to act as a cell death effector in polyglutamine-induced cell death [69, 70].
In addition to the two main pathways that initiate the caspase cascade — namely, the death receptor (extrinsic) pathway, which involves caspase-8 (or caspase-10), and the mitochondrial (intrinsic) pathway, which involves caspase-9 as the apical caspase [71–73] — studies from multiple laboratories point to a caspase-12-mediated intrinsic apoptotic pathway that involves the endoplasmic reticulum and the UPR [42, 74–76, 77, 78]. Caspase-12, which is associated with the ER, is specifically involved in apoptosis that results from ER stress (at least in murine cells; whether the human caspase-12 gene actually encodes an enzymatically active caspase remains controversial [42, 68, 74–76, 77, 79]). Caspase-12 may be activated by m-calpain, a cysteine protease activated by perturbed calcium homeostasis in ER-stressed cells, or by caspase-7, which is recruited to the ER in stressed cells, or by ER-stress-activated IRE1, which recruits caspase-12 through tumor necrosis factor receptor-associated factor 2 (TRAF2) protein [75, 76, 80]. Caspase-12, together with caspase-9, triggers a downstream apoptotic pathway that is independent of Apaf-1, cytochrome c and mitochondria
ER stress, UPR and neurodegeneration
Misfolded proteins, and the associated ER stress, are emerging as virtually constant features of neurodegenerative diseases. That UPR- and ER-stress-induced cell death could be involved in the pathogenesis of several neurodegenerative disorders (as opposed to being causally unrelated correlates) comes from several recent reports. The accumulation of misfolded proteins resulting in alterations in the structure of organelles, including the ER, has been observed in transgenic models of HD, AD and ALS, as well as in huntingtin-null mice [83–99]. Examples of such misfolded proteins, and related complexes, include the neurotoxic oligomers of the Aβ-peptide in AD [100–102]; cytoplasmic inclusions (Lewy bodies) that stain for α-synuclein, parkin and an unfolded form of the PAEL (parkin-associated endothelin receptor-like) protein [2, 102–105] in PD; intracellular inclusions in degenerating neurons and glia that stain for mutant CuZnSOD [83, 106, 107] in murine models of ALS; and the expanded polyglutamine (poly Q) aggregates that trigger ER-stress-induced cell death in HDand other polyglutamine expansion diseases [104, 105, 108, 109, 110, 111]. Since these neurodegenerative diseases may be caused by specific mutant proteins that accumulate as misfolded proteins and escape degradation, it is likely that ER stress plays an important pathogenetic role in these diseases.
Thus recent studies have suggested the following: first, a role for presenilin-1 in the activation of IRE1 and induction of the UPR [112]; second, a role for GRP78 (Bip) in binding and limiting the production of Aβ peptide; and third, reduced cytotoxicity of the β-amyloid peptide in caspase-12 deficient mice, suggesting a link between the role of the UPR and ER stress in AD [75, 113]. In mouse models of AD featuring the overexpression of mutant presenilin-1, the UPR is downregulated and neurons are highly susceptible to ER-stress-induced cell death [114–121]. Neurons expressing PS1 mutations exhibit increased sensitivity to death induced by DNA damage, and the hypersensitivity to DNA damage is correlated with increased intracellular Ca2+ levels, induction of p53, upregulation of the Ca2+-dependent protease m-calpain, mitochondrial membrane depolarization and activation of caspase-12 [114–117]. Another ER protein, HERP (homocysteine-induced ER-stress-responsive protein), regulates PS-mediated Aβ generation and accumulation and the formation of senile plaques and vascular Aβ deposits. These data, taken together, implicate caspases and misfolded or unfolded Aβ in UPR-induced apoptosis [122].
Several lines of transgenic mice engineered to express mutant forms of the CuZnSOD gene develop a motor neuron disease (MND) that resembles human familial amyotrophic lateral sclerosis (FALS). These mice show prominent degeneration of mitochondria and ER in spinal cord neurons [123–126]. Aggregate formation leading to extensive dilation of the ER is also observed in mouse models of ALS featuring the expression of CuZnSOD mutants, and this is not observed with the expression of wild-type CuZnSOD protein [84, 86, 125–127]. Furthermore, in tissue culture models, mutant CuZnSOD (but not wild-type CuZnSOD) aggregates in association with the ER and induces ER-stress-associated increases in the levels of GRP78/BiP [123]. The viability of neurons expressing the mutant CuZnSOD is protected by overexpressing heat-shock-protein 70 (HSP70), arguing that protein folding plays a role in cytotoxicity, at least in this model [128].
PD is characterized pathologically by the loss of dopaminergic neurons, primarily in the substantia nigra pars compacta, and by the presence of ubiquitinated protein deposits in the neuronal cytoplasm (Lewy bodies), as well as by protein inclusions within neurites. These deposits and inclusions contain aggregates of α-synuclein (a small presynaptic protein of incompletely defined function), which is mutated in some of the familial cases of PD [129–133]. In a Drosophila model of PD, overexpression of wild-type or mutant α-synuclein triggers dopaminergic neuron cell loss that is prevented by overexpression of Hsp70 [134]. Similarly, mutations in another protein dubbed parkin — a member of the E3 ubiquitin ligase family of proteins — result in autosomal recessive juvenile parkinsonism (ARJP) [135, 136]. Overexpression of normal parkin inhibits ER-stress-induced cell death triggered by cellular parkin that is mutated in juvenile parkinsonism, suggesting that accumulation of misfolded mutant parkin (or one of the targets of its E3 ligase activity) might contribute to the selective dopaminergic neuronal cell death observed in ARJP [135–137]. One potentially critical target, a putative G-protein-coupled transmembrane polypeptide named PAEL receptor, is a parkin-binding protein, and may be a target for degradation mediated by the E3 ligase activity of parkin. Overexpression of this receptor unfolds the protein, decreasing its solubility, and results in unfolded-protein-induced cell death. A marked increase in PAEL receptor, presumably due to a defect in parkin-mediated degradation, was demonstrated in the brains of ARJP patients [136, 138, 139].
Toxins including rotenone, 6-hydroxydopamine (6-OHDA) and 1-methyl-4-phenylpyridinium (MPP+) cause the death of dopaminergic neurons in vitro and in vivo, and are widely used to model PD. Treatment of dopaminergic cells with these drugs triggers the induction of a large number of genes involved in ER stress and the unfolded protein response, such as ER chaperones and elements of the ubiquitin-proteasome system [140, 141]. These results are also compatible with the notion of a link between PD, the UPR and ER stress.
HD, a fatal autosomal-dominant neurodegenerative disease involving predominantly the caudate nucleus and the cerebral cortex, causes involuntary movements, emotional disturbance and cognitive decline. The exact mechanisms underlying neuronal death in HD are still incompletely defined; however, the molecular basis of HD has been shown to be the polyglutamine (polyQ) expansion in the N terminus of Huntingtin (Htt), a cytosolic protein expressed in almost all cells of the body. Numerous theories have been advanced to explain the selective neurodegeneration in this disease, such as the induction of mitochondrial dysfunction and subsequent excitotoxic injury, oxidative stress and apoptosis. Studies demonstrating the involvement of UPR and ER stress in trinucleotide-repeat disorders have also been reported [142]. The cytoplasmic accumulation of polyQ triggers ER stress, apparently by inhibiting the ubiquitin–proteasome system [108, 110, 111]. Subsequently, ER stress activates both the TRAF2–ASK1 complex and caspase-12-mediated apoptotic pathways [108, 110], and overexpression of Hsp70 suppresses polyQ toxicity [143]. Postmortem brain samples from patients with Huntington’s disease show a selective decrease in ER-associated α-glucosidase and fucosyl-transferase activities in the putamen, suggesting that these changes reflect highly specific alterations in glycoprotein synthesis and processing and may contribute to the underlying pathology of these disorders [144].
Conclusions
The ER is very sensitive to changes in its environment, and such changes may lead to disruption of its normal homeostasis. A variety of environmental insults, as well as genetic diseases associated with the accumulation of misfolded proteins, can all affect the ER structure, function and integrity, leading to ER stress and contributing to the pathogenesis of different disease states. Prolonged stress leads to organelle damage and dysfunction, and ultimately triggers PCD. The accumulation of misfolded proteins seen in various neurodegenerative diseases leads to an ER stress response, irrespective of whether the misfolded proteins build up within the ER or outside the ER. Further insights into the pathways triggered by misfolded proteins, ER stress responses and cell death programs should facilitate the development of new therapeutic strategies for neurodegenerative disorders and other disorders that feature misfolded proteins.
Acknowledgements
Work in this laboratory is supported by the National Institutes of Health (AG12282 and NS45093 to D.E.B, NS33376 to D.E.B. and R.V.R), the Joseph Drown Foundation, and the Institute for the Study of Aging.
Abbreviations
AD = Alzheimer’s disease
ALG-2 = apoptosis-linked gene 2
ALS = amyotrophic lateral sclerosis
ARJP = autosomal recessive juvenile parkinsonism
ATF6 = activating transcription factor 6
eiF2α = eukaryotic translation initiation factor 2 alpha
ER = endoplasmic reticulum
GRP = glucose regulated protein
GRP78/Bip = glucose-regulated protein of 78 kilodaltons
HD = Huntington’s disease
HSP70 = heat-shock-protein 70
IRE-1 = inositol-requiring transmembrane kinase/endoribonuclease
PAEL = parkin-associated endothelin receptor-like
PCD = programmed cell death
PD = Parkinson’s disease
PERK = PKR-like ER kinase
polyQ = polyglutamine
UPR = unfolded protein response
VCP = valosin-containing protein
XBP-1 = X-box-binding protein 1
References:
Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426:900–904
Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296:1991–1995
Kopito RR, Ron D. Conformational disease. Nat Cell Biol. 2000;2:E207–E209
Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890
Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530
Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–1233
Dobson CM, Ellis RJ. Protein folding and misfolding inside and outside the cell. EMBO J. 1998;17:5251–5254
Sitia R, Braakman I. Quality control in the endoplasmic reticulum protein factory. Nature. 2003;426:891–894
Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899
Sherman MY, Goldberg AL. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29:15–32
Ron D. Translational control in the endoplasmic reticulum stress response. J Clin Invest. 2002;110:1383–1388
Ma Y, Hendershot LM. The mammalian endoplasmic reticulum as a sensor for cellular stress. Cell Stress Chaperones. 2002;7:222–229
Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the mammalian unfolded protein response. Annu Rev Cell Dev Biol. 2002;18:575–599
Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. 2004;14:20–28
Paschen W, Frandsen A. Endoplasmic reticulum dysfunction — a common denominator for cell injury in acute and degenerative diseases of the brain? J Neurochem. 2001;79:719–725
Harding HP, Ron D. Endoplasmic reticulum stress and the development of diabetes: a review. Diabetes. 2002;51:S455–S461
Lee AS. Mammalian stress response: induction of the glucose-regulated protein family. Curr Opin Cell Biol. 1992;4:267–273
Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded- protein response. Nat Cell Biol. 2000;2:326–332
Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, Wek RC. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol Cell Biol. 1998;18:7499–7509
Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274
Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904
Liu CY, Schroder M, Kaufman RJ. Ligand-independent dimerization activates the stress response kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. J Biol Chem. 2000;275:24881–24885
Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6:1355–1364
Chen X, Shen J, Prywes R. The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J Biol Chem. 2002;277:13045–13052 This paper, together with [23,25], describes the phenomenon of regulated intracellular proteolysis. The luminal domain of ATF6 senses ER stress and translocates to the Golgi, where it is processed by two enzymes that process SREBPs in response to cholesterol deprivation. The cleaved fragment translocates to the nucleus and activates transcription of the endogenous GRP78/BiP gene. The results show that ATF6 has a mechanism to sense ER stress and that, in addition to their involvement in lipid synthesis, S1P and S2P are required for the ER stress response.
Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–3799
Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, Yoshida H, Mori K, Kaufman RJ. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002;16:452–466 See annotation to [27••].
Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891 A very thorough study by these two groups [26••,27••] describing the two-step activation of XBP-1 that includes the transcriptional activation by ATF6 and post-transcriptional activation by Ire1. Both reports suggest that IRE1α and ATF6 signaling pathways merge through regulation of XBP1 activity to induce downstream gene expression for full activation of the UPR.
Li M, Baumeister P, Roy B, Phan T, Foti D, Luo S, Lee AS. ATF6 as a transcription activator of the endoplasmic reticulum stress element: thapsigargin stress-induced changes and synergistic interactions with NF-Y and YY1. Mol Cell Biol. 2000;20:5096–5106
Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, Prywes R. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J Biol Chem. 2000;275:27013–27020
Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002;3:99–111
Sidrauski C, Walter P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell. 1997;90:1031–1039
Kawahara T, Yanagi H, Yura T, Mori K. Endoplasmic reticulum stress-induced mRNA splicing permits synthesis of transcription factor Hac1p/Ern4p that activates the unfolded protein response. Mol Biol Cell. 1997;8:1845–1862
Patil C, Walter P. Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr Opin Cell Biol. 2001;13:349–355
Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96
Liu CY, Xu Z, Kaufman RJ. Structure and intermolecular interactions of the luminal dimerization domain of human IRE1α J Biol Chem. 2003;278:17680–17687
Ma K, Vattem KM, Wek RC. Dimerization and release of molecular chaperone inhibition facilitate activation of eukaryotic initiation factor-2 kinase in response to endoplasmic reticulum stress. J Biol Chem. 2002;277:18728–18735
Hendershot LM, Wei JY, Gaut JR, Lawson B, Freiden PJ, Murti KG. In vivo expression of mammalian BiP ATPase mutants causes disruption of the endoplasmic reticulum. Mol Biol Cell. 1995;6:283–296
Morris JA, Dorner AJ, Edwards CA, Hendershot LM, Kaufman RJ. Immunoglobulin binding protein (BiP) function is required to protect cells from endoplasmic reticulum stress but is not required for the secretion of selective proteins. J Biol Chem. 1997;272:4327–4334
Xiao G, Chung TF, Pyun HY, Fine RE, Johnson RJ. KDEL proteins are found on the surface of NG108–15 cells. Brain Res Mol Brain Res. 1999;72:121–128
Triantafilou M, Fradelizi D, Triantafilou K. Major histocompatibility class one molecule associates with glucose regulated protein (GRP) 78 on the cell surface. Hum Immunol. 2001;62:764–770
Delpino A, Castelli M. The 78 kDa glucose-regulated protein (GRP78/BIP) is expressed on the cell membrane, is released into cell culture medium and is also present in human peripheral circulation. Biosci Rep. 2002;22:407–420
Rao RV, Peel A, Logvinova A, del Rio G, Hermel E, Yokota T, Goldsmith PC, Ellerby LM, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program: role of the ER chaperone GRP78. FEBS Lett. 2002;514:122–128 See annotation to [43•].
Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J Biol Chem. 2003;278:20915–20924 These reports [42•,43•] describe the localization of GRP78 as a trans-membrane ER protein and elaborate on its role in blocking caspase-dependent cell death.
Lee AS. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem Sci. 2001;26:504–510
Rudner J, Jendrossek V, Belka C. New insights in the role of Bcl-2 Bcl-2 and the endoplasmic reticulum. Apoptosis. 2002;7:441–447
Germain M, Shore GC. Cellular distribution of Bcl-2 family proteins. Sci STKE. 2003;2003:pe10
Thomenius MJ, Wang NS, Reineks EZ, Wang Z, Distelhorst CW. Bcl-2 on the endoplasmic reticulum regulates Bax activity by binding to BH3-only proteins. J Biol Chem. 2003;278:6243–6250
Granville DJ, Carthy CM, Jiang H, Shore GC, McManus BM, Hunt DW. Rapid cytochrome c release, activation of caspases 3, 6, 7 and 8 followed by Bap31 cleavage in HeLa cells treated with photodynamic therapy. FEBS Lett. 1998;437:5–10
Maatta J, Hallikas O, Welti S, Hilden P, Schroder J, Kuismanen E. Limited caspase cleavage of human BAP31. FEBS Lett. 2000;484:202–206
Ng FW, Shore GC. Bcl-XL cooperatively associates with the Bap31 complex in the endoplasmic reticulum, dependent on procaspase-8 and Ced-4 adaptor. J Biol Chem. 1998;273:3140–3143
Ito Y, Pandey P, Mishra N, Kumar S, Narula N, Kharbanda S, Saxena S, Kufe D. Targeting of the c-Abl tyrosine kinase to mitochondria in endoplasmic reticulum stress-induced apoptosis. Mol Cell Biol. 2001;21:6233–6242
Reimertz C, Kogel D, Rami A, Chittenden T, Prehn JH. Gene expression during ER-stress-induced apoptosis in neurons: induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J Cell Biol. 2003;162:587–597
Bourdon JC, Renzing J, Robertson PL, Fernandes KN, Lane DP. Scotin, a novel p53-inducible proapoptotic protein located in the ER and the nuclear membrane. J Cell Biol. 2002;158:235–246
Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135–139 See annotation to [55••].
Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730 These two reports [54••,55••] describe how cells lacking both Bax and Bak are resistant to multiple apoptotic stimuli, including the ER stress stimuli thapsigargin and tunicamycin. The cells have a reduced resting concentration of calcium in the ER and several apoptotic stimuli required both ER-released Ca2+ and the presence of mitochondrial BAX or BAK to fully restore apoptosis. Thus BAX and BAK are required for triggering apoptotic signals both in the ER and mitochondria.
Nutt LK, Chandra J, Pataer A, Fang B, Roth JA, Swisher SG, O’Neil RG, McConkey DJ. Bax-mediated Ca2+ mobilization promotes cytochrome c release during apoptosis. J Biol Chem. 2002;277:20301–20308
Nutt LK, Pataer A, Pahler J, Fang B, Roth J, McConkey DJ, Swisher SG. Bax and Bak promote apoptosis by modulating endoplasmic reticular and mitochondrial Ca2+ stores. J Biol Chem. 2002;277:9219–9225
Vander Heiden MG, Thompson CB. Bcl-2 proteins: regulators of apoptosis or of mitochondrial homeostasis? Nat Cell Biol. 1999;1:E209–E216
Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J, Thompson CB. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol. 2003;162:59–69
Annis MG, Zamzami N, Zhu W, Penn LZ, Kroemer G, Leber B, Andrews DW. Endoplasmic reticulum localized Bcl-2 prevents apoptosis when redistribution of cytochrome c is a late event. Oncogene. 2001;20:1939–1952
Hacki J, Egger L, Monney L, Conus S, Rosse T, Fellay I, Borner C. Apoptotic crosstalk between the endoplasmic reticulum and mitochondria controlled by Bcl-2. Oncogene. 2000;19:2286–2295
Wang XZ, Lawson B, Brewer JW, Zinszner H, Sanjay A, Mi LJ, Boorstein R, Kreibich G, Hendershot LM, Ron D. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153) Mol Cell Biol. 1996;16:4273–4280
Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–1108
Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995
McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–1259
Breckenridge DG, Nguyen M, Kuppig S, Reth M, Shore GC. The procaspase-8 isoform, procaspase-8L, recruited to the BAP31 complex at the endoplasmic reticulum. Proc Natl Acad Sci USA. 2002;99:4331–4336
Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol. 2003;160:1115–1127
Rao RV, Poksay KS, Castro-Obregon S, Schilling B, Row RH, Del Rio G, Gibson BW, Ellerby HM, Bredesen DE. Molecular components of a cell death pathway activated by endoplasmic reticulum stress. J Biol Chem. 2004;279:177–187
Kobayashi T, Tanaka K, Inoue K, Kakizuka A. Functional ATPase activity of p97/valosin-containing protein (VCP) is required for the quality control of endoplasmic reticulum in neuronally differentiated mammalian PC12 cells. J Biol Chem. 2002;277:47358–47365
Hirabayashi M, Inoue K, Tanaka K, Nakadate K, Ohsawa Y, Kamei Y, Popiel AH, Sinohara A, Iwamatsu A, Kimura Y, et al. VCP/p97 in abnormal protein aggregates, cytoplasmic vacuoles, and cell death, phenotypes relevant to neurodegeneration. Cell Death Differ. 2001;8:977–984
Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell. 1997;91:443–446
Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem. 1999;68:383–424
Nijhawan D, Honarpour N, Wang X. Apoptosis in neural development and disease. Annu Rev Neurosci. 2000;23:73–87
Nakagawa T, Yuan J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol. 2000;150:887–894
Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103
Rao RV, Hermel E, Castro-Obregon S, del Rio G, Ellerby LM, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Mechanism of caspase activation. J Biol Chem. 2001;276:33869–33874
Rao RV, Castro-Obregon S, Frankowski H, Schuler M, Stoka V, Del Rio G, Bredesen DE, Ellerby HM. Coupling endoplasmic reticulum stress to the cell death program. An Apaf-1-independent intrinsic pathway. J Biol Chem. 2002;277:21836–21842 See annotation to [78•].
Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic-reticulum-stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem. 2002;277:34287–34294 The data from both papers [77•,78•] indicate that the intrinsic mitochondrial pathway may not be essential during ER-stress-induced cell death.
Saleh M, Vaillancourt JP, Graham RK, Huyck M, Srinivasula SM, Alnemri ES, Steinberg MH, Nolan V, Baldwin CT, Hotchkiss RS, et al. Differential modulation of endotoxin responsiveness by human caspase-12 polymorphisms. Nature. 2004;429:75–79. 1476–4687
Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, Tohyama M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem. 2001;276:13935–13940
Oubrahim H, Chock PB, Stadtman ER. Manganese(II) induces apoptotic cell death in NIH3T3 cells via a caspase-12-dependent pathway. J Biol Chem. 2002;277:20135–20138
Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K, et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Aβ-induced cell death. J Cell Biol. 2004;165:347–356
Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819
Dal Canto MC, Gurney ME. Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am J Pathol. 1994;145:1271–1279
Guidetti P, Charles V, Chen EY, Reddy PH, Kordower JH, Whetsell WO, Jr, Schwarcz R, Tagle DA. Early degenerative changes in transgenic mice expressing mutant huntingtin involve dendritic abnormalities but no impairment of mitochondrial energy production. Exp Neurol. 2001;169:340–350
Higgins CM, Jung C, Ding H, Xu Z. Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J Neurosci. 2002;22:RC215
Hilditch-Maguire P, Trettel F, Passani LA, Auerbach A, Persichetti F, MacDonald ME. Huntingtin: an iron-regulated protein essential for normal nuclear and perinuclear organelles. Hum Mol Genet. 2000;9:2789–2797
Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, et al. A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192
Iannicola C, Moreno S, Oliverio S, Nardacci R, Ciofi-Luzzatto A, Piacentini M. Early alterations in gene expression and cell morphology in a mouse model of Huntington’s disease. J Neurochem. 2000;75:830–839
Julien JP. Amyotrophic lateral sclerosis. unfolding the toxicity of the misfolded. Cell. 2001;104:581–591
Mattson MP, Chan SL, Camandola S. Presenilin mutations and calcium signaling defects in the nervous and immune systems. Bioessays. 2001;23:733–744
Lyons TJ, Liu H, Goto JJ, Nersissian A, Roe JA, Graden JA, Cafe C, Ellerby LM, Bredesen DE, Gralla EB, et al. Mutations in copper-zinc superoxide dismutase that cause amyotrophic lateral sclerosis alter the zinc binding site and the redox behavior of the protein. Proc Natl Acad Sci USA. 1996;93:12240–12244
Martindale D, Hackam A, Wieczorek A, Ellerby L, Wellington C, McCutcheon K, Singaraja R, Kazemi-Esfarjani P, Devon R, Kim SU, et al. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat Genet. 1998;18:150–154
Nasir J, Floresco SB, O’Kusky JR, Diewert VM, Richman JM, Zeisler J, Borowski A, Marth JD, Philips AG, Hayden MR. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81:811–823
Reddy PH, Williams M, Tagle DA. Recent advances in understanding the pathogenesis of Huntington’s disease. Trends Neurosci. 1999;22:248–255
Tabrizi SJ, Workman J, Hart PE, Mangiarini L, Mahal A, Bates G, Cooper JM, Schapira AH. Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann Neurol. 2000;47:80–86
Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW. Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington’s disease. Proc Natl Acad Sci USA. 2000;97:8093–8097
White JK, Auerbach W, Duyao MP, Vonsattel JP, Gusella JF, Joyner AL, MacDonald ME. Huntingtin is required for neurogenesis and is not impaired by the Huntington’s disease CAG expansion. Nat Genet. 1997;17:404–410
Wiedau-Pazos M, Goto JJ, Rabizadeh S, Gralla EB, Roe JA, Lee MK, Valentine JS, Bredesen DE. Altered reactivity of superoxide dismutase in familial amyotrophic lateral sclerosis. Science. 1996;271:515–518
Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxic effects of amyloid β protein: reversal by tachykinin neuropeptides. Science. 1990;250:279–282
Glabe C. Intracellular mechanisms of amyloid accumulation and pathogenesis in Alzheimer’s disease. J Mol Neurosci. 2001;17:137–145
Martin JB. Molecular basis of the neurodegenerative disorders. N Engl J Med. 1999;340:1970–1980
Xu J, Kao SY, Lee FJ, Song W, Jin LW, Yankner BA. Dopamine-dependent neurotoxicity of α-synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat Med. 2002;8:600–606
Kakizuka A. Protein precipitation: a common etiology in neurodegenerative disorders? Trends Genet. 1998;14:396–402
Kakizuka A. Recent progress in the research field on triplet repeat diseases. Rinsho Shinkeigaku. 1998;38:988–992
Williamson TL, Corson LB, Huang L, Burlingame A, Liu J, Bruijn LI, Cleveland DW. Toxicity of ALS-linked SOD1 mutants. Science. 2000;288:399
Rothstein JD, Dykes-Hoberg M, Corson LB, Becker M, Cleveland DW, Price DL, Culotta VC, Wong PC. The copper chaperone CCS is abundant in neurons and astrocytes in human and rodent brain. J Neurochem. 1999;72:422–429
Kouroku Y, Fujita E, Jimbo A, Kikuchi T, Yamagata T, Momoi MY, Kominami E, Kuida K, Sakamaki K, Yonehara S, et al. Polyglutamine aggregates stimulate ER stress signals and caspase-12 activation. Hum Mol Genet. 2002;11:1505–1515
Matsuzawa A, Nishitoh H, Tobiume K, Takeda K, Ichijo H. Physiological roles of ASK1-mediated signal transduction in oxidative stress- and endoplasmic-reticulum-stress-induced apoptosis: advanced findings from ASK1 knockout mice. Antioxid Redox Signal. 2002;4:415–425 In this work involving ASK1 KO mice, the authors demonstrate that ASK1?/? primary neurons are defective in polyQ-, proteasome-inhibitor-, and ER-stress-induced JNK activation and cell death. Their findings suggest that ASK1 is a key element in ER-stress-induced cell death and may play an important role in the neuropathological alterations seen in polyQ diseases.
Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–1355
Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555
Niwa M, Sidrauski C, Kaufman RJ, Walter P. A role for presenilin-1 in nuclear accumulation of Ire1 fragments and induction of the mammalian unfolded protein response. Cell. 1999;99:691–702
Yang Y, Turner RS, Gaut JR. The chaperone BiP/GRP78 binds to amyloid precursor protein and decreases Aβ40 and Aβ42 secretion. J Biol Chem. 1998;273:25552–25555
Terro F, Czech C, Esclaire F, Elyaman W, Yardin C, Baclet MC, Touchet N, Tremp G, Pradier L, Hugon J. Neurons overexpressing mutant presenilin-1 are more sensitive to apoptosis induced by endoplasmic reticulum-Golgi stress. J Neurosci Res. 2002;69:530–539
Chan SL, Culmsee C, Haughey N, Klapper W, Mattson MP. Presenilin-1 mutations sensitize neurons to DNA damage-induced death by a mechanism involving perturbed calcium homeostasis and activation of calpains and caspase-12. Neurobiol Dis. 2002;11:2–19
Katayama T, Imaizumi K, Honda A, Yoneda T, Kudo T, Takeda M, Mori K, Rozmahel R, Fraser P, George-Hyslop PS, et al. Disturbed activation of endoplasmic reticulum stress transducers by familial Alzheimer’s disease-linked presenilin-1 mutations. J Biol Chem. 2001;276:43446–43454
Katayama T, Imaizumi K, Sato N, Miyoshi K, Kudo T, Hitomi J, Morihara T, Yoneda T, Gomi F, Mori Y, et al. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat Cell Biol. 1999;1:479–485
Keller JN, Guo Q, Holtsberg FW, Bruce-Keller AJ, Mattson MP. Increased sensitivity to mitochondrial toxin-induced apoptosis in neural cells expressing mutant presenilin-1 is linked to perturbed calcium homeostasis and enhanced oxyradical production. J Neurosci. 1998;18:4439–4450
Kudo T, Katayama T, Imaizumi K, Yasuda Y, Yatera M, Okochi M, Tohyama M, Takeda M. The unfolded protein response is involved in the pathology of Alzheimer’s disease. Ann N Y Acad Sci. 2002;977:349–355
Mattson MP, Gary DS, Chan SL, Duan W. Perturbed endoplasmic reticulum function, synaptic apoptosis and the pathogenesis of Alzheimer’s disease. Biochem Soc Symp. 2001;67:151–162
Paschen W, Doutheil J. Disturbances of the functioning of endoplasmic reticulum: a key mechanism underlying neuronal cell injury? J Cereb Blood Flow Metab. 1999;19:1–18
Sai X, Kawamura Y, Kokame K, Yamaguchi H, Shiraishi H, Suzuki R, Suzuki T, Kawaichi M, Miyata T, Kitamura T, et al. Endoplasmic reticulum stress-inducible protein, Herp, enhances presenilin-mediated generation of amyloid β-protein. J Biol Chem. 2002;277:12915–12920
Tobisawa S, Hozumi Y, Arawaka S, Koyama S, Wada M, Nagai M, Aoki M, Itoyama Y, Goto K, Kato T. Mutant SOD1 linked to familial amyotrophic lateral sclerosis, but not wild-type SOD1, induces ER stress in COS7 cells and transgenic mice. Biochem Biophys Res Commun. 2003;303:496–503
Shibata N. Transgenic mouse model for familial amyotrophic lateral sclerosis with superoxide dismutase-1 mutation. Neuropathology. 2001;21:82–92
Tu PH, Gurney ME, Julien JP, Lee VM, Trojanowski JQ. Oxidative stress, mutant SOD1, and neurofilament pathology in transgenic mouse models of human motor neuron disease. Lab Invest. 1997;76:441–456
Mourelatos Z, Gonatas NK, Stieber A, Gurney ME, Dal Canto MC. The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu,Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc Natl Acad Sci USA. 1996;93:5472–5477
Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116
Bruening W, Roy J, Giasson B, Figlewicz DA, Mushynski WE, Durham HD. Up-regulation of protein chaperones preserves viability of cells expressing toxic Cu/Zn-superoxide dismutase mutants associated with amyotrophic lateral sclerosis. J Neurochem. 1999;72:693–699
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047
Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat Genet. 1998;18:106–108
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-synuclein in Lewy bodies. Nature. 1997;388:839–840
Conway KA, Lee SJ, Rochet JC, Ding TT, Harper JD, Williamson RE, Lansbury PT., Jr Accelerated oligomerization by Parkinson’s disease linked α-synuclein mutants. Ann N Y Acad Sci. 2000;920:42–45
Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4:1318–1320
Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of α-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science. 2002;295:865–868 This work highlights the important roles played by chaperones, in this case Hsp70, in preventing dopaminergic neuronal loss associated with α-synuclein, suggesting that chaperones may play a role in PD progression.
Imai Y, Soda M, Takahashi R. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J Biol Chem. 2000;275:35661–35664
Takahashi R, Imai Y, Hattori N, Mizuno Y. Parkin and endoplasmic reticulum stress. Ann N Y Acad Sci. 2003;991:101–106
Farrer M, Chan P, Chen R, Tan L, Lincoln S, Hernandez D, Forno L, Gwinn-Hardy K, Petrucelli L, Hussey J, et al. Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol. 2001;50:293–300
Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell. 2001;105:891–902
Takahashi R, Imai Y. Pael receptor, endoplasmic reticulum stress, and Parkinson’s disease. J Neurol. 2003;250(Suppl 3):III25–III29
Holtz WA, O’Malley KL. Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J Biol Chem. 2003;278:19367–19377
Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson’s disease. J Neurosci. 2002;22:10690–10698
Mattson MP. Excitotoxic and excitoprotective mechanisms: abundant targets for the prevention and treatment of neurodegenerative disorders. Neuromolecular Med. 2003;3:65–94
Warrick JM, Paulson HL, Gray-Board GL, Bui QT, Fischbeck KH, Pittman RN, Bonini NM. Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell. 1998;93:939–949
Cross AJ, Crow TJ, Johnson JA, Dawson JM, Peters TJ. Loss of endoplasmic reticulum-associated enzymes in affected brain regions in Huntington’s disease and Alzheimer-type dementia. J Neurol Sci. 1985;71:137–143.

Return to ALZHEIMER's
Since 4-12-2020
| Home Page | Visit Our Sponsors | Become a Sponsor |
Please read our DISCLAIMER |