

Sulforaphane - Role in Aging and Neurodegeneration This section is compiled by Frank M. Painter, D.C.
Send all comments or additions to: Frankp@chiro.org


FROM: Geroscience 2019 (Oct); 41 (5): 655–670 ~ FULL TEXT
Roberto Santín-Márquez, Adriana Alarcón-Aguilar, Norma Edith López-Diazguerrero, Niki Chondrogianni, Mina Königsberg
Departamento de Ciencias de la Salud,
División de Ciencias Biológicas y de la Salud,
Universidad Autónoma Metropolitana-Iztapalapa,
A.P. 55-535, 09340, Mexico City, Mexico.
In the last several years, numerous molecules derived from plants and vegetables have been tested for their antioxidant, anti-inflammatory, and anti-aging properties. One of them is sulforaphane (SFN), an isothiocyanate present in cruciferous vegetables. SFN activates the antioxidant and anti-inflammatory responses by inducing Nrf2 pathway and inhibiting NF-κB. It also has an epigenetic effect by inhibiting HDAC and DNA methyltransferases and modifies mitochondrial dynamics. Moreover, SFN preserves proteome homeostasis (proteostasis) by activating the proteasome, which has been shown to lead to increased cellular lifespan and prevent neurodegeneration. In this review, we describe some of the molecular and physical characteristics of SFN, its mechanisms of action, and the effects that SFN treatment induces in order to discuss its relevance as a "miraculous" drug to prevent aging and neurodegeneration.
Keywords: HDAC; NF-κB; Nrf2; Oxidative stress; Proteasome.
From the FULL TEXT Article:
Sulforaphane
Discovery and isolation
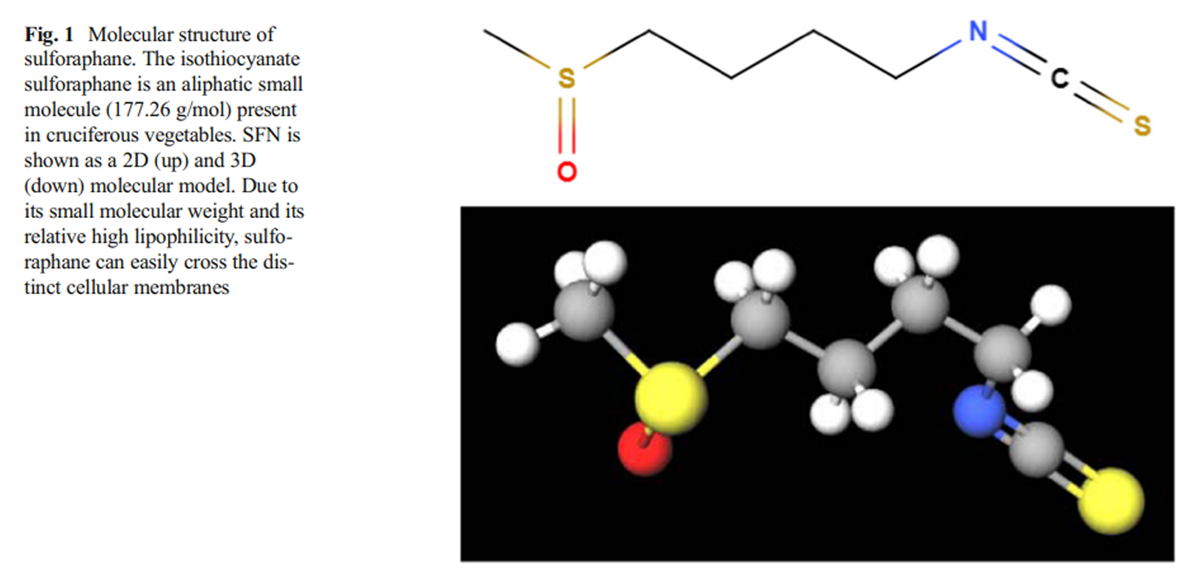
Figure 1 Since 1980s, it was known that the consumption of green and yellow vegetables (leafy green vegetables, cauliflower, carrots, broccoli, Brussel sprouts, etc.) was related to a reduction in the risk of developing certain types of cancer (Colditz et al. 1985). Later, studies showed that some molecules contained in those vegetables were capable to induce xenobiotic metabolism and antioxidant enzyme activation and, as a common feature, those molecules were glutathione S-transferase (GST) substrates, becoming potential protective agents against developing cancer (Talalay et al. 1988). Later, different green and yellow vegetables were tested to determine which of those molecules could activate phase II enzyme activity, with the cruciferous family being the one that enhanced these enzymatic activities the most, particularly broccoli and Brussel sprout extracts (Prochaska et al. 1992). Almost immediately, an isothiocyanate described as “a potent phase II enzymes inducer” was isolated from broccoli extracts, and was identified by spectroscopic methods as-1-isothiocyanato-(4R)-(methylsulfinyl) butane or sulforaphane (SFN); (Zhang et al. 1992). Phase II enzymes are composed of antioxidant and conjugating enzyme which are capable of enhancing xenobiotic hydrophilicity, facilitating their excretion, promoting cell detoxification and antioxidant response (Xu et al. 2005). Following the publication of these results, many studies were focused in the SFN synthesis, mechanism of action, and its protective role against tumorigenesis and electrophile attacks provoked by oxidative stress (Prestera et al. 1993; Talalay et al. 1995) (Figure 1).
Biosynthesis and other products after glucoraphanin
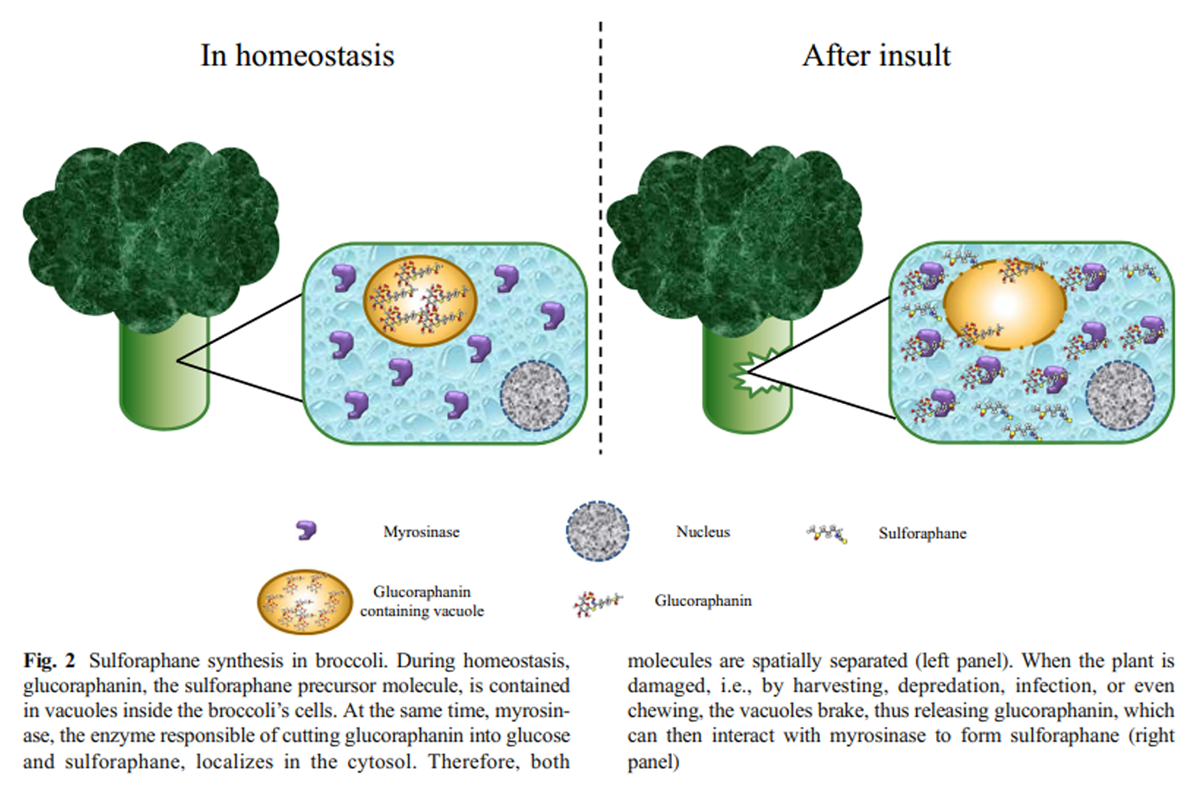
Figure 2 SFN is the hydrolysis product of glucoraphanin (4-(methylsulfinyl) butyl glucosinolate), one of the main glucosinolates contained in cruciferous vegetables, and the most abundant in broccoli and Brussel sprouts (Ghawi et al. 2013; Brown et al. 2002). Glucosinolates are a family of amino acid–derived secondary metabolites characterized by the presence of three moieties: sulfur groups within a thiohydroximate-O-sulfonate structure; a D-glucose molecule; and an α-amino acid–derived alkyl, aralkyl, or indolyl side chain (Barba et al. 2016; Ishida et al. 2014).
The glucoraphanin biosynthetic pathway comprises, at least, three stages:(1) a side chain elongation round by deamination of amino acids to a 2-oxo-acid;
(2) glucosinolate core structure formation by S-glucosyltransferases; and
(3) a S-oxygenation mediated by monooxygenases, to finally get the bioactive form of glucoraphanin as product (Ishida et al. 2014; Yang et al. 2017).The glucoraphanin concentration in the plant mostly depends on the organ, post-germination time, environmental conditions, post-harvest handling conditions, and storage (Guo et al. 2014; Winkler et al. 2007; Martinez-Villaluenga et al. 2010). When the plant containing glucosinolates is mechanically damaged (cut, chewed, or chopped), or is under stress conditions, such as a bacterial or fungal infection, a hydrolase called myrosinase (thioglucoside glucohydrolase), which is usually separated in a different compartment, is released to interact with glucoraphanin molecules to hydrolyze it, releasing a D-glucose molecule and a thiohydroximate-O-sulfate, an unstable aglycone (Burmeister et al. 2000), which spontaneously rearranges to the isothiocyanate form of sulforaphane (Angelino and Jeffery 2014; Barba et al. 2016; Bones and Rossiter 2006) (Figure 2).
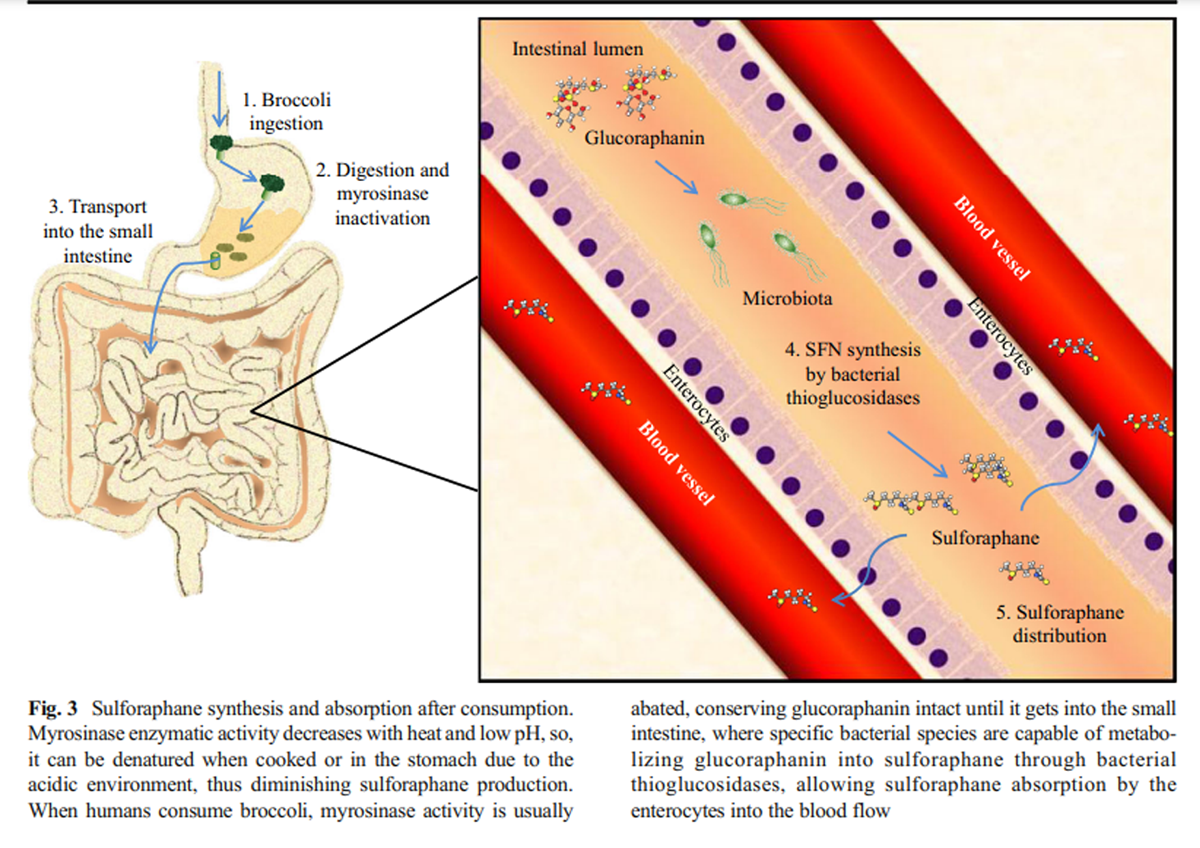
Figure 3 Myrosinase activity decreases with heat exposure when the vegetables are cooked before eating, favoring the presence of glucoraphanin and diminishing the availability of sulforaphane when the vegetables are cooked before consumption (Ghawi et al. 2013). However, myrosinase activity is not only found in plants, it could also be found an equivalent activity in mammal’s lower gut microbiota thioglucosidases, allowing the hydrolysis of glucoraphanin and the sulforaphane absorption (Lai et al. 2010; Bheemreddy and Jeffery 2007). Some species of bacteria, such as Escherichia coli, Bacteroides thetaiotaomicron, Enterococcus faecalis, Enterococcus faecium, Peptostreptococcus sp., and Bifidobacterium sp. can process glucosinolates due to the presence of specific thioglucosidases, maintaining the glucoraphanin conversion to isothiocyanates even after myrosinase heat deactivation (Hullar and Fu 2014) (Figure 3).
Physicochemical characterization
Sulforaphane is a small molecule with a molecular weight of 177.28 g/mol and its molecular formula is C6H11NOS2 with a melting point between 58.6 and 91.2 °C (Fishbein and Heilman 2018). Although glucoraphanin is hydrophilic, sulforaphane, like other isothiocyanates, is typically an aliphatic lipophilic molecule (Fahey and Talalay 1999).
Pharmacokinetics
Absorption, distribution, metabolism, and elimination
Due to sulforaphane’s small molecular weight and its relatively high lipophilicity, it is rapidly absorbed in the jejunum across the enteric cells after oral administration (Petri et al. 2003). It reaches the highest concentrations in plasma 3 h after consumption (approximately 0.9 μmol/L), and slowly decreases after the second hour, having an approximate half-life of 2.2 h (Hanlon et al. 2008; Hu et al. 2004; Cramer and Jeffery 2011). When SFN is in the cells, it is metabolized by phase II and III enzymes, for example, glutathione S-transferase (GST) to form conjugated products such as sulforaphane-glutathione (SFN-GSH), sulforaphane-cysteine (SFN-Cys), sulforaphane N-acetyl cysteine (SFN-NAC), and sulforaphane-cysteinyl-glycine (SFN-CG), which are thought to be important for the activation of several SFN biological effects (Clarke et al. 2011). The primary sulforaphane metabolism sites are the intestinal walls; the liver, where it is conjugated with GSH; the kidney, where it is conjugated with NAC; and the bladder (Verkerk et al. 2009) and is accumulated mainly in those same organs, and in lower concentrations in plasma, skin, and lung tissues (Bricker et al. 2014). The excretion rate is higher at the 6th hour after administration (Atwell et al. 2015), excreting in urine SFN-NAC as a principal metabolite, and total elimination is reached in the 12th hour post-ingestion (Cramer and Jeffery 2011).
Proposed mechanisms of action
Keap1/Nrf2-dependent mechanism
Given that SFN was recognized as a potent inductor of xenobiotic metabolism and antioxidant response, the Keap1/Nrf2 pathway was a very attractive candidate as SFN target. Under basal redox conditions, the nuclear factor erythroid 2-related factor 2 (Nrf2) is sequestered by a Kelch-like ECH-associated protein 1 (Keap1) dimer and rapidly ubiquitinated by the recruitment of the E3 ubiquitin ligase Cul3, resulting in Nrf2 degradation via the proteasome (Li et al. 2004; Silva-Palacios et al. 2018). When the redox state becomes predominantly oxidized, some regulatory cysteines of Keap1 (such as cys151, cys273, and cys288) are oxidized and promote a conformational change that facilitates Nrf2 release and prevents its degradation (Dinkova-Kostova et al. 2017). After being released, Nrf2 requires certain post-translational modifications, namely, tyrosine or serine phosphorylation by different kinases, such as MAPK or PKC that phosphorylate Nrf2 in Tyr568, as well as the kinase GSK-3β, which in turn phosphorylates Nrf2 in Ser40. Both phosphorylations are related to Nrf2 nuclear translocation (Bhakkiyalakshmi et al. 2015). When Nrf2 is imported into the nucleus, it forms a heterodimer with small Maf proteins (MafG, MafK, MafF), which endows it with a DNA-binding capacity to attach to its consensus sequence, the antioxidant response element (ARE; Silva-Palacios et al. 2018), resulting in the transcription of diverse antioxidant response genes, such as NAD(P) H quinone dehydrogenase 1 (NQO1), heme oxygenase 1 (HO-1), and γ-glutamylcysteine ligase (γ GCL), among many others (Guerrero-Beltrán et al. 2012).
In vivo experiments have demonstrated an increased Nrf2 expression and nuclear localization after SFN treatment (Bai et al. 2013), as well as an augmented transcriptional activity (Pu et al. 2018; Zhao et al. 2016). It has also been proposed that SFN is able to oxidize the regulatory cys151 in the Keap1 dimer, thus stopping Nrf2 degradation (Hu et al. 2011).
Interestingly, the Nrf2-dependent antioxidant response is diminished during aging (Zhou et al. 2018), but the SFN treatment has been shown to increase Nrf2 transcription, activation, nuclear translocation, DNA-binding, and antioxidant gene expression in epithelial cells isolated from old rats and elderly humans (Kubo et al. 2017). Therefore, SFN has been proven to be an important inducer of the antioxidant and protective response during aging.
Keap1/Nrf2-independent mechanismsNF-κB mechanism Although SFN administration has been mainly related to the antioxidant response, it has been demonstrated that it is also involved in the regulation of the inflammatory response via the NF-κB pathway. NF-κB pathway is considered to be the classical signaling for the inflammatory process and it is present in all cell types. This pathway is involved in the transcription and regulation of different pro- and anti-inflammatory mediators, depending on the activator stimuli. NF-κB can be activated by two different pathways: the “canonical” and the “alternative,” keeping in common the presence of different inhibitors from the IκB family (transcriptional repressors that halt NF-κB in the cytosol), and the IκB kinase family (IKK; proteins involved in the IκB phosphorylation and subsequent degradation; (Lawrence 2009; Shih et al. 2011; Sun 2017). In mammals, the NF-κB transcription factor family consists of five distinct proteins: p65 (RelA), RelB, c-Rel, p105/p50 (NF-κB1), and p100/52 (NF-κB2), which can form both heterodimers and homodimers, with p50/p65 heterodimer being the most abundant (Oeckinghaus and Ghosh 2009).
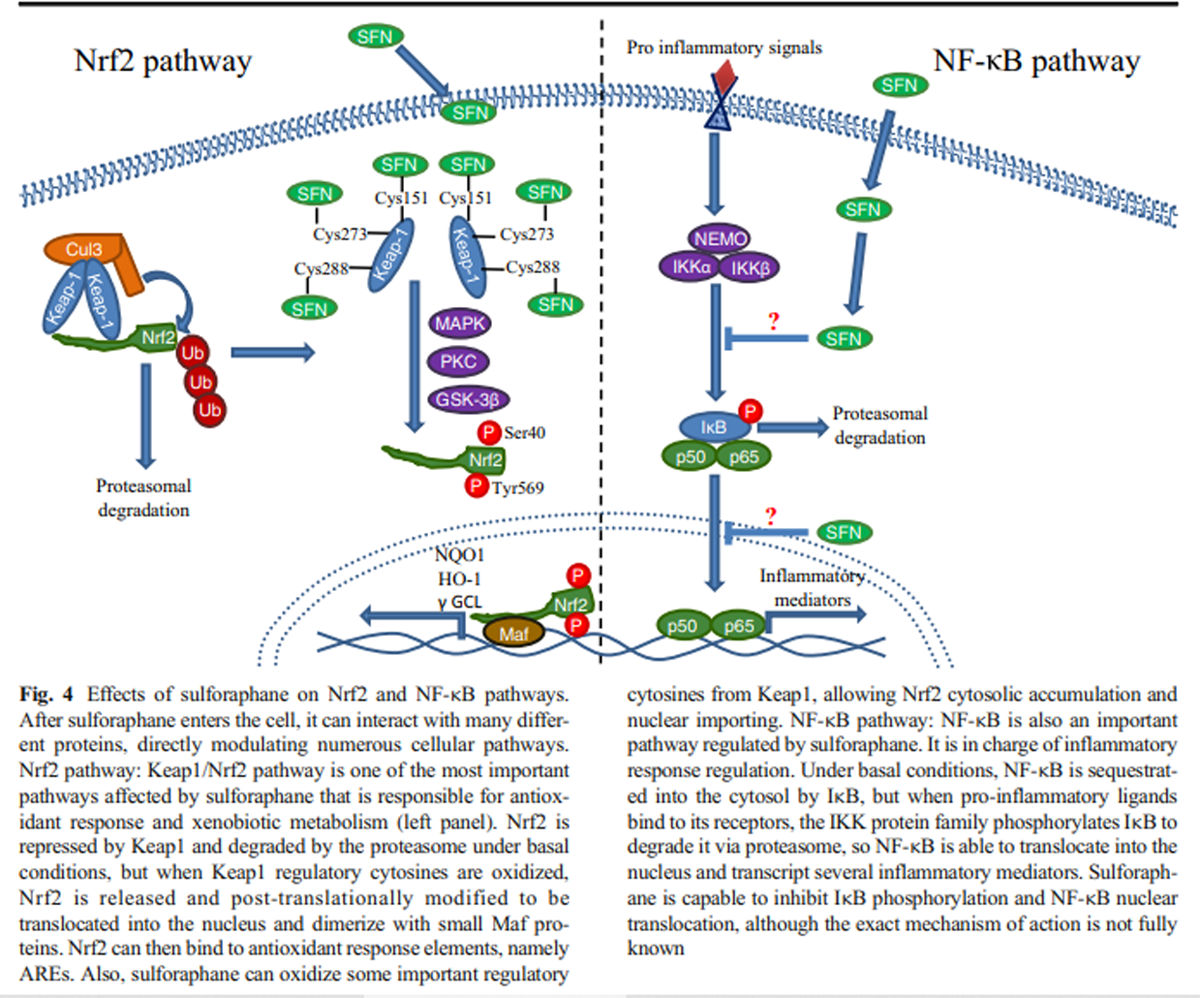
Figure 4 Although it is not fully elucidated how SFN inhibits the inflammatory response, it is known that SFN is capable to diminish NF-κB nuclear translocation and DNA-binding capacity (Heiss et al. 2001). Inflammation master regulators, like TNF-α or IL-6, also decrease after SFN treatment, suggesting an inhibition of the NF-κB pathway (Negi et al. 2011). Even the secretion of several pro-inflammatory cytokines such as IL-2, IL-4, IL-6, and IFN-γ is inhibited in a SFN dose-dependent manner (Checker et al. 2015), suggesting that NF-κB could be directly interacting with SFN. However, the effect of this isothiocyanate in the inflammatory response can be observed upon its effect on other proteins different from NF-κB, such as IKB, the NF-κB repressor responsible for its cytosolic localization, whose degradation is inhibited in a dose-dependent manner in vivo after SFN treatment (Nallasamy et al. 2014) (Figure 4).
Epigenetic mechanism Another mechanism of action for SFN was proposed several years ago by Myzac, Ho, and Dashwood. Knowing that SFN-NAC (SFN-N-Acetylcysteine) and SFN-Cys are important metabolites formed when SFN is metabolized through the mercapturic acid pathway, and using a molecular modeling approach, they discovered a plausible interaction for the carboxylate group of SFN-Cys within the active site of HDACs (histone deacetylases) using Zn as a bidentate ligand (Myzak et al. 2004). This interaction of SFN metabolites inhibits the HDAC activity, thus altering the cellular epigenetic pathways.
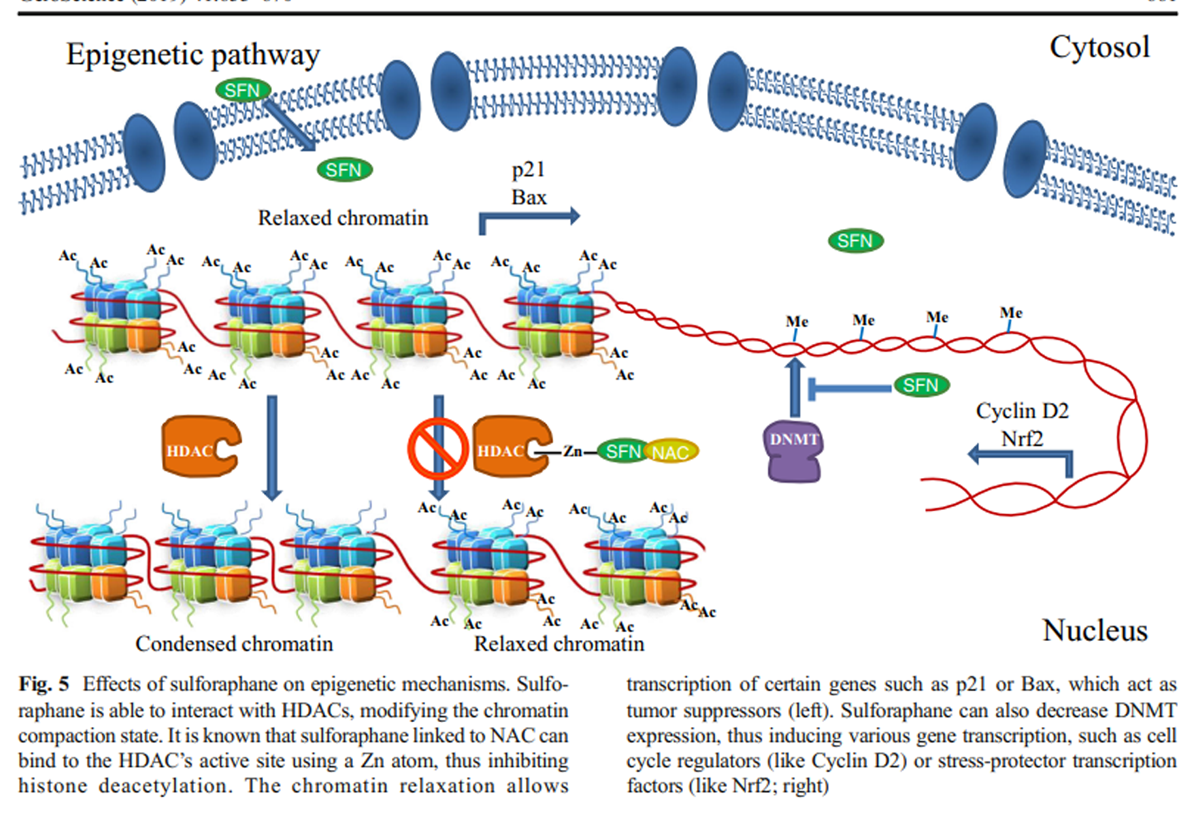
Figure 5 The same research group established an association between HDAC inhibition and histone acetylation increase on Bax and p21 promoters (Myzak et al. 2006b); Bax is a well-known antiapoptotic protein that belongs to the Bcl2 family, while p21 is a cell cycle inhibitor that binds to CDK-cyclins and blocks their effect. So, elevated Bax and p21 mRNA and protein expression have been associated with cell cycle arrest and apoptotic cell death in cancer cells after SFN treatment due to an epigenetic mechanism (Myzak et al. 2006a, 2006b) (Figure 5).
Currently, it is also known that this particular isothiocyanate and its metabolites decrease the expression of DNA methyltransferases (DNMTs), especially DNMT1 and DNMT3b. In particular, SFN diminishes methylation in cyclin D2 promoter regions containing c-Myc and multiple Sp1 binding sites (Hsu et al. 2011). These results, together with SFN HDAC inhibitory effect, place these molecules as important epigenetic regulators, which are able to induce the transcriptional activation of several tumor suppressor genes (Dashwood and Ho 2008).
Being SFN, an isothiocyanate derived from cruciferous vegetables; it has become an important object of investigation as a functional food or as a food supplement. To date, there are numerous studies where SFN improves cellular conditions due to its epigenetic role.
The effect of SFN as an HDAC inhibitor was mainly explored to explain its anti-tumorigenic effect using essentially human embryonic kidney 293 cells, HCT116 human colorectal cancer cells and BPH-1, LnCaP and PC-3 prostate epithelial cells, and also some in vivo murine models (Myzak et al. 2006b), where SFN was shown to increase the acetylation of histone H3 and histone H4.
SFN is also known to inhibit to TERT expression by decreasing DNMTs in MCF-7 and MDA-MB-231 human breast cancer cells (Meeran et al. 2010); therefore, SFN alone or in combination with other natural compounds such as genistein (Paul et al. 2018), withaferin A (Royston et al. 2017), or epigallocatechin-3-gallate (Li et al. 2016) inhibits cellular proliferation of breast cancer cell lines.
Even though there are many reports related to SFN’s neuroprotective effect in brain cells and neurodegenerative diseases, most of them are related to its effects associated to Nrf2 activation, which were already discussed above. Nevertheless, there are very few studies addressing SFN’s epigenetic mechanism in the brain. More specifically, Zhao et al. (2018) found that SFN augmented the antioxidant and anti-inflammatory capacity of mouse neuroblastoma N2a cells that overexpress the human Swedish mutant amyloid precursor protein (N2a/APPswe cells), which is a common cellular model of Alzheimer’s disease. They found that SFN upregulated Nrf2 expression by reducing DNA demethylation levels of the Nrf2 promoter (Zhao et al. 2018), in a very interesting study that involves both the epigenetic mechanism and the Nrf2-activation mechanism. In another model using the triple-transgenic mouse model of Alzheimer’s disease (3 × Tg-AD), Kim et al. (2017) showed that the use of SFN regulates the expression of the Brain-derived neurotrophic factor (BDNF) via HDAC inhibition, thus increasing H3 and H4 acetylation on the BDNF promoter. Enhancing BDNF expression as an effect of SFN treatment increased the neuronal content of several synaptic molecules like MAP 2, synaptophysin, and PSD-95 in primary cortical neurons of 3 × Tg-AD, suggesting that this molecule might be an attractive drug to minimize neurodegenerative disorders. However, more research is needed to evaluate the epigenetic mechanism of SFN in the brain.
Sulforaphane effects on proteasome status
Proteasome in aging and neurodegeneration
Proteasomes are the main cellular degradation machineries implicated in almost all pathways and (normal and pathological) conditions through their role in the maintenance of proteome homeostasis (proteostasis; Papaevgeniou and Chondrogianni 2016). The main eukaryotic proteasome complex is called 20S core that is produced by the association of 4 rings composed by 7 different α- and β-type subunits in an α7β7β7α7 order. On one or both edges of the 20S core, various regulatory complexes may be bound giving rise to higher molecular weight complexes with the 26S/30S proteasome being the most studied (produced through binding of one (26S) or two (30S) 19S complexes at the 20S core ends) (Ciechanover 1998; Budenholzer et al. 2017).
Apart of the role of this multi-enzyme in the regulation of almost all vital cellular processes such as the cell cycle and protein catabolism, it is also in charge of the cellular detoxification from oxidized and misfolded proteins that accumulate during the progression of aging or age-related diseases (Chondrogianni et al. 2015). We, among others, have shown that the proteasome function is reduced during the progression of cellular senescence and aging (Chondrogianni et al. 2000, 2003, 2008). Consequently, proteasome activation has emerged as a hot spot in the anti-aging field (Chondrogianni et al. 2015b; Vilchez et al. 2014). More specifically, proteasome activation in primary fibroblasts either through genetic overexpression of proteasome subunits (Chondrogianni et al. 2005; Chondrogianni and Gonos 2007; Hwang et al. 2007) or through natural compound treatment (Katsiki et al. 2007; Kapeta et al. 2010; Kwak et al. 2007) has been shown to lead to increased cellular lifespan and enhanced stress resistance. The same was shown to occur in lens epithelial cells, thus showing that it is not a cell type-specific effect (Liu et al. 2007). More recently, we achieved proteasome activation in the multicellular organismal level (C. elegans) through genetic overexpression of pbs-5 proteasome subunit (Chondrogianni et al. 2015a) as well as through a natural compound that is a diet constituent (Papaevgeniou et al. 2016). On top of the promotion of proteasome activation on the anti-aging field, this strategy is also emerging in the field of aggregation-related diseases (such as Alzheimer’s, Parkinson’s, Huntington’s disease among others) where the proteasome is either inhibited or downregulated upon the disease progression (Vilchez et al. 2014). Overexpression of Rpn6 proteasome subunit in C. elegans or Rpn11 proteasome subunit in D. melanogaster results in reduced toxicity of poly-Q aggregates and suppressed neurodegeneration in HD models (Vilchez et al. 2012; Park et al. 2009). We have also shown that proteasome activation exerts a protective role against Aβ toxicity in various AD models of C. elegans and in murine primary neurons (Papaevgeniou et al. 2016).
Sulforaphane enhances the proteasomal function
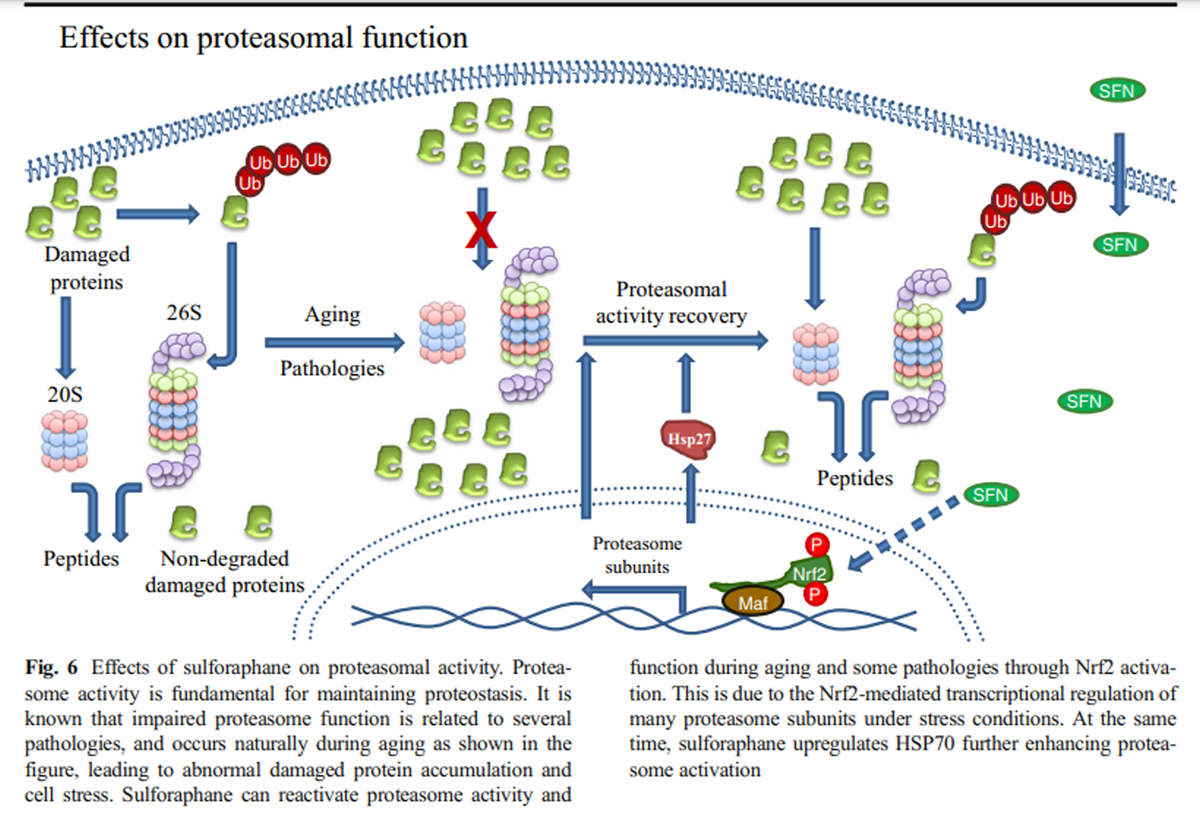
Figure 6 Microarray analysis has shown that most of the proteasome subunits are transcriptional targets of Nrf2 in mammals (Kwak et al. 2003) while the same was also shown for its orthologue namely SKN-1 in C. elegans (Kahn et al. 2008). This regulation occurs under specific conditions such as the cellular response to oxidative stress induced by SFN. In addition to the enhanced SFN-mediated induction of several proteasome subunits (Kwak et al. 2007), increased proteasome activity was also attributed to the SFN-mediated upregulation of Hsp27 that was shown to promote proteasome activation as well (Gan et al. 2010) (Figure 6).
Treatment of murine neuroblastoma cells with SFN enhanced both the expression and the function of proteasome via Nrf2-mediated regulation endowing cells with enhanced resistance against H2O2-mediated oxidative stress and toxicity (Kwak et al. 2007). Additionally, SFN-mediated Nrf2 induction protected neuroblastoma cells from Aβ-induced cell death. This protection was shown to be dependent on proteasome activation promoted by Nrf2 (Park et al. 2009). Upon oxidative stress adaptation, Nrf2-dependent enhancement of oxidized protein degradation has been attributed to the enhanced expression of both the 20S core and the 11S regulator (Pickering et al. 2012) (Fig. 6).
The proteasome activating effects of SFN were also verified in UPS function reporter mice. On top of the proteasome activation, enhancement of the autophagy activity was also revealed in the brain and the liver of the relative mice, suggesting that this might be another pathway to be explored due to SFN treatment.
Sulforaphane effects on the central nervous system
Sulforaphane in neuroinflammation
Neuroinflammation is a common feature of neurological diseases. Inflammasomes, which form a multiprotein complex within the innate immune system, induce inflammation in response to various stimuli, such as pathogens and stress. Inflammasomes activate pro-inflammatory caspases, like caspase-1, that lead to the activation of pro-inflammatory cytokines, including the interleukins IL-1b, IL-18, and IL-33, which promote neuroinflammation and brain pathologies (Pennisi et al. 2017). The toll-like receptors (TLR4) also participate in the process of neuroinflammation, due to ROS and cytokine generation (Iizumi et al. 2016). Both astroglia and microglia express TLR4 receptors, and endogenous ligands produced in the ischemic brain induce inflammatory responses. It has been reported that hyperammonemia induces neuroinflammation and increases GABAergic secretion. Hernandez-Rabaza et al. (2016) showed that SFN administration to Wistar male rats with hyperammonemia decreased IL-1b and GABA, and increased IL-4 and IL-10. Moreover, it has also been observed that in 2-month-old C57BL/6 mice, in which cognitive functions were decreased due to neuroinflammation induced with LPS for 7 days, pre-treatment with SFN 30 min before LPS administration improved the temporal space behavior and the memory compared to those animals administered only with LPS (Gao et al. 2018).
The most recognized idea to explain the mechanism by which SFN decreases neuroinflammation is the activation of the transcription factor Nrf2 and the increase in the expression of its target genes, as described previously. For example, in the microglial cell line BV2 and in the primary microglial cultures from adult and aged mice stimulated with LPS to induce an inflammatory state, SFN treatment decreased the expression of pro-inflammatory markers such as IL-1b, IL-6, and iNOS via Nrf2 antioxidant response (Townsend and Johnson 2016). In an okadaic acid model (OKA) (Tonoki et al. 2009), which induces oxidative stress and neuroinflammation by decreasing Nrf2 levels, the administration of SFN prevented the memory impairment induced by OKA in rats. That treatment also restored Nrf2 and antioxidant protein (GCLC, HO-1) expression, diminished the oxidative stress by attenuating ROS and NO levels, and increased GSH concentration. In addition, neuroinflammation was also diminished by reducing NF-κB and TNF-α, and by rising IL-10 levels, which was followed by a reduction in neuronal apoptosis in the rat’s cerebral cortex and hippocampus (Dwivedi et al. 2016).
Conversely, in a dietary intervention experiment, old mice were fed with a 10% broccoli diet for 28 days, and the neuroinflammation was reduced by an increment of interleukin-1β mRNA and a decrement of b-245 β, preventing the upregulation of reactive glia markers. In this way, a 10% broccoli diet provided a modest reduction in age-related oxidative stress and glial reactivity, but was insufficient to inhibit LPS-induced inflammation (Townsend and Johnson 2016).
Another interesting mechanism proposed to explain SFN participation in the reduction of neuroinflammation is through the inhibition of the transcription factor NF-κB. In a model of human macrophages (THP-1) exposed to the peptide Aβ1-42 to induce an inflammatory state and subsequently treated with SFN, IL-1b and TNF-α levels decreased, and that correlated with a reduction in NF-κB translocation (Jhang et al. 2018). These results suggest that the effect of SFN might be through a combined mechanism of Nrf2 activation and NF-κB inhibition in a situation where the cellular redox state could be playing a preponderant role.
Sulforaphane in the treatment of different neuropathologies
Most of the neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (Bricker et al. 2014), Huntington’s disease (Sachdeva et al. 2014), and multiple sclerosis, are characterized by a compromised redox state, where a deficiency in the antioxidant response, mediated by the transcription factor Nrf2, has generally been observed (Liddell 2017). Since oxidative stress and neuroinflammation have been mainly related to the decrease in cognitive function, it is not surprising that SFN treatment has been used as a conceivable therapy to improved cognitive functions in different animal models.
Several studies have shown that SFN treatment protects against oxidative stress induced by various molecules such as MPTP, 6-OH dopamine, and rotenone in animal models of PD, by increasing Nrf2 activation and regulating the antioxidant response (Jazwa et al. 2011; Morroni et al. 2013; Zhou et al. 2016).
In an HD model using mice treated with 3-NP, SFN treatment decreased the levels of pro-inflammatory cytokines, such as TNF-α, IL-6, iNOS, COX2, as well as NF-κB activation, and increased the Keap-1/Nrf2/ARE pathway (Jang and Cho 2016). Treatment of cells expressing the mutated huntingtin protein (mHtt) with SFN resulted in increased mHtt degradation that was accompanied by decreased mHtt cytotoxicity. This positive effect was abolished upon treatment with MG132, a proteasome inhibitor, thus further demonstrating the tight interplay between SFN and proteasome activity discussed above (Liu et al. 2014).
Multiple sclerosis (MS) is a chronic disease characterized by focal lesions, axon demyelination, and inflammation that involve the infiltration of peripheral macrophages into the central nervous system. MS is a difficult pathology to study in animal models, so it is usually studied by evaluating the effects of autoimmune responses that resemble MS. The effect of SFN on experimental autoimmune encephalomyelitis (EAE) in C57BL/6 mice significantly inhibited the development and severity of this disease, accompanied by reduced spinal cord inflammatory infiltration and demyelination. In addition, the protection induced by SFN was also related to decrease oxidative stress levels in the mice brains through activation of the Nrf2/ARE pathway and the increase in the HO-1 and NQO1 expression. On the other hand, treatment with SFN inhibited the immune responses mediated by the specific Th17 reactions and augmented the anti-inflammatory response mediated by IL-10 (Li et al. 2013).
In the transgenic PS1V97L mice model of AD, 6-month-old animals treated with SFN for 4 months improved their cognitive function in comparison to aged match non-treated animals (Hou et al. 2018). In another AD model, the double transgenic amyloid APP/PS1 (precursor protein/presenilin 1) mice, SFN treatment improved the ability of independent exploration in the open field, the environmental adaptability, and ameliorated the Morris maze test (Zhang et al. 2017). Interestingly, consumption of glucoraphanin, a precursor of SFN, by 6-week-old young mice prevented the cognitive function decline during adulthood when phencyclidine (an NMDA receptor antagonist) was administrated, suggesting that SFN might also be used as a prophylactic treatment to prevent cognitive deficit (Shirai et al. 2015).
More recently, the beneficial effects of SFN on hippocampal-dependent spatial memory impairment induced by post-natal proteasome inhibition was revealed. More specifically, SFN administration was accompanied by Nrf2 nuclear translocation and led to upregulation of β5 proteasome subunit and thus to a potential enhancement of proteasome function that attenuated the deleterious effects of post-natal proteasome inhibition. Mice pretreated with SFN were rescued of spatial memory impairment during adulthood that was developed in mice treated with the proteasome inhibitor MG132 in the absence of SFN. Additionally, SFN treatment promoted the expression of various molecules that are involved in synaptic plasticity (Sunkaria et al. 2018). Protective effects in the brain against stroke were also shown to occur after SFN preconditioning in the cerebral vasculature, although the proteasome status was not examined under those conditions (Alfieri et al. 2013). Nevertheless, the possible therapeutic potential of Nrf2 inducers in vascular disease and aging through the crosstalk of Nrf2 with the proteasome has been suggested (Chapple et al. 2012).
Concerning ALS, there are no conclusive results whether SFN counteracted the dysfunctional effects of the disease; however, it has been observed a slight restoration in Nrf2 levels, which might be an interesting start to treat this pathology (Liddell 2017).
Effects of SFN on aging
Longevity and lifespan
Currently, aging is considered a multifactorial, universal, progressive, and deleterious process that occurs over time in living beings, and that has as a consequence the decline in the organism’s physiological functions, increasing their vulnerability to disease and death (López-Otín et al. 2013; Organization, WHO 2015).
Several studies have reported an increment in oxidative stress and a decay in antioxidant defense mechanisms during aging. This situation decreases the organism’s protection against numerous oxidative, chemical, and pathological stresses (Dai et al. 2014; Poprac et al. 2017), since it has been proposed that redox status might function as a mediator between the genome and epigenome (genes expression) and the exposome (environment, diet, life style, etc. (Jones 2015).
One of the features that is known to decrease during aging is the Nrf2 signaling (Sachdeva et al. 2014; Zhang et al. 2015; Zhou et al. 2018). However, some bioactive molecules, like SFN, are able to re-activate the Nrf2 pathway through all the mechanisms discussed above, thus associating SFN treatment with the prevention of aging decline and age-related pathologies (Greco et al. 2011). Nevertheless, the concentrations of SFN used must be taken into consideration, because it is known that SFN has different effects depending on the cell type and organism, an aspect that is still being studied in the aging context.
In a human model of cataracts associated to aging, lens epithelial cells (hLECs) are known to accumulate oxidative damage by decreasing the antioxidant enzymes, particularly a reduction in Prdx6. When those cells were treated with SFN, they increased the Nrf2 activity leading to Prdx6 expression and cytoprotection against UVB-induced injury (Kubo et al. 2017).
Several studies suggest that aging is the result of an age-associated decline in stem cell self-renewal, replication, and lineage commitment (Sharpless and DePinho 2007). When the effects of SFN were evaluated on the biology of human mesenchymal stem cells (MSCs), it was found that low doses of SFN promoted MSC proliferation and protected them from apoptosis and senescence, but higher doses had a cytotoxic and contrary effect. Probably, the adverse effects of SFN were due to its pro-oxidant function generating glutathione depletion and superoxide production (Zanichelli et al. 2012).
It is well known that increased cell senescence contributes to aging-associated diseases. Senescence can be induced by stressors like oxidative stress, dysfunctional telomeres, excessive mitogen signaling, perturbations in chromatin organization, oxidative DNA damage, and oncogenic activation, among others (Campisi and di Fagagna 2007; Childs et al. 2015). Many studies have shown a continuous increase in glucose metabolism through glycolysis in senescence. Hariton et al. (2018) showed evidence that treatment with 1 μM SFN once per week delayed the senescence of human MRC-5 and BJ fibroblasts, exhibiting a caloric restriction mimetic-like activity and a decreased oxidative damage to proteins and DNA.
As mentioned above, SFN modulates several epigenetic modifications such as DNA methylation and histone modification (Meeran et al. 2010). It was shown that SFN participates in the inhibition/modulation of HDAC and DNMT activity leading to the reactivation of epigenetically silenced genes in order to enhance chemoprevention (Tortorella et al. 2015). These changes in the epigenome aid in the prevention of neoplasms, resulting in cancer cell death, and potentiate the longevity effects of caloric restriction (Daniel and Tollefsbol 2015).
Regarding premature aging, lately there is an increase interest on studying the protection conferred by SFN against cellular skin aging. Skin aging is sensitive to stress factors including ultraviolet radiation (UVR), excessive alcohol consumption, tobacco abuse, and environmental pollution that lead to cumulative deterioration in skin appearance and function (Pontius and Smith 2011). SFN has demonstrated protective effects against ultraviolet-induced skin damage through several mechanisms of action, by a decrement of oxidative stress (Talalay et al. 2007), and maintenance of collagen levels during photo-aging via the inhibition of the AP-1 activation and the expression of metalloproteinases (Zhu et al. 2004).
Related to the effect of SFN on longevity, in a study of beetles fed with flour diet supplemented with lyophilized broccoli, elongation of their lifespan under physiological conditions (32 °C) as well as during heat-stress conditions (42 °C) was observed. Broccoli also increased longevity in the absence of stress, with the most significant benefit achieved using 1–5% broccoli. The transcription factor Nrf2 increased stress resistance by induction of detoxification, regulation of key stress-resistant factors such as Nrf-2, Jnk-1, and Foxo-1 that resulted to longevity (Grünwald et al. 2013).
The Hutchinson-Gilford progeria syndrome (HGPS) recapitulates the normal aging process, with the expression of mutated lamin A known as ‘Progerin’ causing DNA damage and genomic instability (Gordon et al. 2014). Progerin is also known to alter various components of the cellular proteolytic pathways, thus leading to impaired proteasome activity and autophagy. Accordingly, the beneficial effects of SFN on HGPS were also revealed. Fibroblasts from patients and normal individuals were treated with sulforaphane, and progerin clearance was induced (Gabriel et al. 2015). On top of that, the authors showed that long-term treatment of both normal and HGPS fibroblasts with sulforaphane resulted in increased proliferation rates. This was in agreement with the pro-longevity effects of another Nrf2 activator, namely 18α-glycyrrhetinic acid that was shown to act as pro-longevity factor both in normal fibroblasts (Kapeta et al. 2010) and in C. elegans (Papaevgeniou et al. 2016).
Conclusions
In summary, SFN has been shown to modulate several cellular pathways in order to activate diverse protective responses, which might allow avoiding cancer and neurodegeneration as well as improving cellular lifespan and health span. However, several interesting questions that still remain unanswered are: Does sulforaphane activate all Nrf2 target genes or is there a selectivity for particular ones such as antioxidant or proteasome genes? If so, how is this selection done? Could SFN be used as an anti-aging compound? Might the Nrf2 induction and NF-κB repression lead to side effects? What concentrations might be used in cells and animals and for how long? Does SFN have the same effect on young animals as it does on old animals?
Answers to those questions will eventually reveal the therapeutic potential of Nrf2 inducers in general but also the relative protective potential of sulforaphane.
Funding information
This work was supported by CONACyT grant FON.INST/298/2016, as well as the “Red Temática de Investigación en Salud y Desarrollo Social” from CONACYT, Mexico. Santín-Márquez is a CONACyT scholarship holder. Research from NC lab is currently co-financed by the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, (a) under the call RESEARCH – CREATE – INNOVATE (project code: T1EDK-00353 and T1EDK-01610) and (b) under the Action “Action for the Strategic Development on the Research and Technological Sector” (project STHENOS-b, MIS 5002398).
References:Refer to the Full-Text article 
Return to SULFORAPHANE
Since 7-09-2023
| Home Page | Visit Our Sponsors | Become a Sponsor |
Please read our DISCLAIMER |