

Netrin-1 Interacts with Amyloid Precursor Protein
and Regulates Amyloid-beta ProductionThis section is compiled by Frank M. Painter, D.C.
Send all comments or additions to: Frankp@chiro.org


FROM: Cell Death Differ. 2009 (May); 16 (5): 655–663 ~ FULL TEXT
FC Lourenço, V Galvan, J Fombonne, V Corset, F Llambi, U Müller, DE Bredesen, and P Mehlen
Apoptosis, Cancer and Development Laboratory -
Equipe labellisée La Ligue,
CNRS UMR5238, Centre Léon Bérard,
University of Lyon,
Lyon, France.
The beta-amyloid precursor protein (APP) is an orphan transmembrane receptor whose physiological role is largely unknown. APP is cleaved by proteases generating amyloid-beta (Abeta) peptide, the main component of the amyloid plaques that are associated with Alzheimer's disease. Here, we show that APP binds netrin-1, a multifunctional guidance and trophic factor. Netrin-1 binding modulates APP signaling triggering APP intracellular domain (AICD)-dependent gene transcription.
Furthermore, netrin-1 binding suppresses Abeta peptide production in brain slices from Alzheimer model transgenic mice. In this mouse model, decreased netrin-1 expression is associated with increased Abeta concentration, thus supporting netrin-1 as a key regulator of Abeta production. Finally, we show that netrin-1 brain administration in Alzheimer model transgenic mice may be associated with an amelioration of the Alzheimer's phenotype.
KEYWORDS: Alzheimer’s disease, netrin-1, amyloid-β, amyloid precursor protein
From the FULL TEXT Article:
Background
Alzheimer’s disease (AD), the most common form of dementia, is a progressive neurodegenerative disorder characterized by extracellular deposits of Aβ peptide in senile plaques, intraneuronal neurofibrillary tangles, synapse loss, and cognitive decline. [1] It is widely believed that the accumulation of Aβ, a small peptide with a high propensity to form oligomers and aggregates, is central to the pathogenesis of AD. Aβ derives from the proteolytic cleavage of the transmembrane protein, APP. [2] Although a considerable amount is known about interacting proteins and processing events for APP, the physiological role(s) of APP and its related family members, APLP1 and APLP2 (amyloid precursor-like proteins 1 and 2), is (are) still poorly understood. [2, 3] APP has been proposed to function in cell adhesion and motility, as well as synaptic transmission and plasticity (for a review, see Turner et al. [3]).
The cloning and characterization of APP revealed that it possesses many features reminiscent of a membrane-anchored receptor. However, to date, no clear candidate has emerged as the major ligand triggering APP-mediated signal transduction (although several molecules have been shown to bind APP, such as collagen (types I and IV), heparan sulfate proteoglycan, laminin, glypican, and more recently TAG-1) [4–7] – at least in part because the signal transduction mediated by APP remains incompletely understood. Here, we show that netrin-1 functions as a ligand for APP, it modulates APP signaling, and we also show that it regulates Aβ peptide production in Alzheimer model transgenic mice.
Netrin-1 is a soluble molecule initially described by Tessier-Lavigne and colleagues8 as a chemotropic cue involved in axon guidance. Netrin-1 plays a critical role during nervous system development by mediating chemoattraction of axons and neurons through its interaction with DCC (deleted in colorectal cancer). [8–10] However, netrin-1 has also been described as a survival factor, involved in, among other effects, the regulation of tumorigenesis. [11] Such dual effects on nervous system development and tumorigenesis are characteristics of the so-called dependence receptors, of which DCC represents an example. [12]
While performing a mass spectrometric analysis to identify proteins interacting with either DCC or with the DCC/netrin-1 complex, we sequenced APLP1 as a candidate coimmuno-precipitant with the DCC/netrin-1 complex (Supplementary Figure 1). This led to the question of whether netrin-1 may actually interact with APP family members. We show here that APP is a functional receptor for netrin-1. We also show that netrin-1 negatively regulates the Aβ level in adult brain and that netrin-1 brain administration may represent an appealing strategy to improve the Alzheimer’s phenotype.
Results and Discussion
Netrin-1 interacts with APP
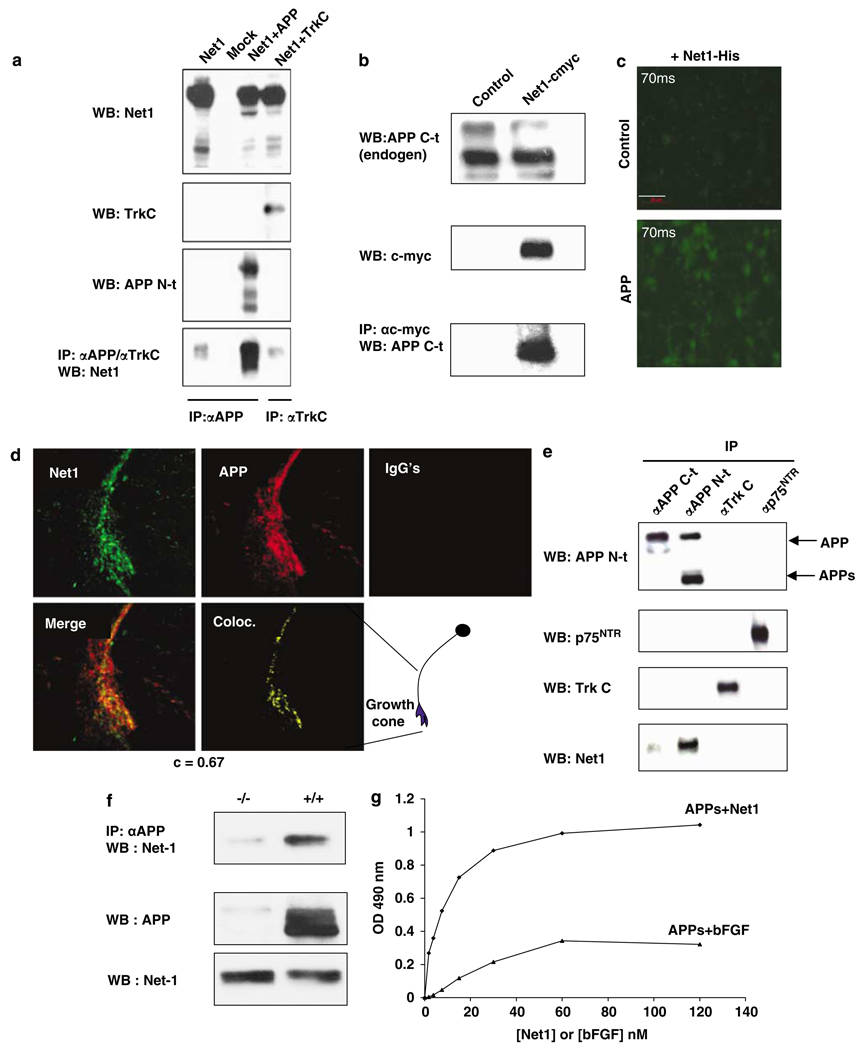
Figure 1 APP or APLP-1 and netrin-1 were coexpressed in HEK293T cells and, as shown in Figure 1a and Supplementary Figure 2ab, APLP1 and APP coimmunoprecipitated with netrin-1. As a negative control, another transmembrane receptor, TrkC, failed to coimmunoprecipitate with netrin-1 (Figure 1a). Similar results were obtained when netrin-1 and APP were coexpressed in B103 cells (data not shown). Not only ectopically expressed APP (Figure 1a) but also endogenous APP could be immunoprecipitated with netrin-1 in HEK293T cells (Figure 1b). We next analyzed whether netrin-1 could be recruited to the plasma membrane of APP-expressing B103 cells. As shown in Figure 1c, although netrin-1 failed to show affinity for control-transfected B103 cells, it was recruited to APP-transfected B103 cells. Because these assays were performed in cells in which APP or netrin-1 were expressed in a nonphysiological setting, we investigated the putative APP/netrin-1 interaction in primary cortical neurons from E16.5 mouse embryos. We first analyzed whether endogenous APP is colocalized with endogenously expressed netrin-1 by confocal microscopy. As shown in Figure 1d, APP colocalizes with netrin-1, especially in growth cones of these cortical neurons. We then performed the coimmunoprecipitation of endogenous proteins. As shown in Figure 1e, netrin-1 interacts with APP in the developing cortex, whereas in the same setting, netrin-1 fails to interact with p75NTR. Moreover, in APP mutant developing brains, netrin-1 is not pulled down after APP immunoprecipitation (Figure 1f). Thus, endogenous netrin-1 specifically interacts with endogenous APP in the developing brain.
To further analyze whether APP interaction is restricted to netrin-1, we performed immunoprecipitation using different netrin family members. In addition to netrin-1, netrin-2 was also found to interact with APP, whereas we failed to detect the interaction of APP with the more divergent netrin molecule, netrin G1 (Supplementary Figure 2c). Because both netrins and APP are heparin-binding proteins, [10, 13] we assessed whether a netrin-1 mutant deleted for domain C (which contains the major heparin-binding domain, but is dispensable for netrin-1 function [10]) retains the ability to interact with APP. As shown in Supplementary Figure 2c, this mutant netrin-1 does indeed retain the ability to interact with APP.
To exclude the possibility of an indirect interaction between netrin-1 and APP, direct in vitro interaction was assessed by immunoprecipitation and ELISA assays on recombinant αAPPs or DCC ectodomain, with recombinant netrin-1 or bFGF, a molecule that shares many characteristics of netrin-1. [14] As shown in Figure 1g and Supplementary Figure 2d, immunoblots and ELISA assays revealed a specific interaction of APP with netrin-1, whereas bFGF/APP failed to show specific binding. The affinity of netrin-1 for APPs is the same order of magnitude as its affinity for DCC (estimated KdAPP/netrin of 6 nM, compared to the known KdDCC/netrin–1 of 10nM [10]). Taken together, these data support the notion that netrin-1 interacts with APP with an affinity that is similar to that of its previously described physiological interaction with DCC.
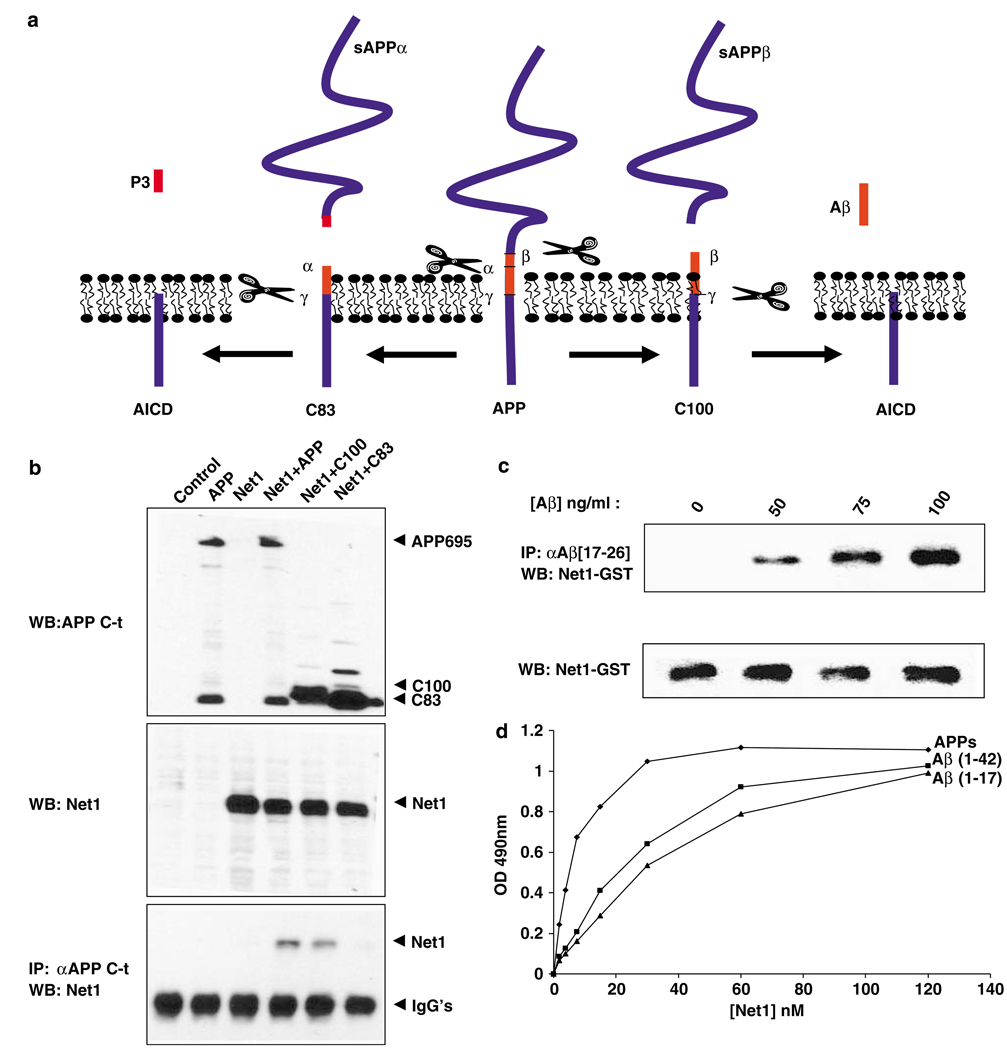
Figure 2 We next attempted to define the APP domain required for the APP/netrin-1 interaction. The carboxy-terminal C83 derived from the γ-secretase cleavage of APP fails to interact with netrin-1, hence further showing that netrin-1 interacts with APP ectodomain (Figures 2a and b). Interestingly, the C100 protein, derived from the γ-secretase cleavage of APP (see scheme in Figure 2a), is pulled down with netrin-1 (Figure 2b). Taken together with the finding that, in vitro, the αAPPs protein interacts with netrin-1 (Figure 1j and Supplementary Figure 2b), these observations suggested that a binding region of APP is localized between the β-cleavage and the α-cleavage sites – that is, the 17 aminoterminal residues of Aβ, even though this may not be the only interacting domain of APP with netrin-1. Along this line, recombinant netrin-1 interacts in a concentration-dependent manner with the Aβ peptide (Figure 2c). Thus, Aβ appears to interact with netrin-1 (although we cannot exclude the possibility that this interaction is biologically irrelevant and only because of the well-described “sticky” nature of the Aβ peptide). Interestingly, not only Aβ but also a smaller fragment of Aβ, Aβ 1–17 – that is, the 17 first amino acids of Aβ (a less toxic peptide than full-length Aβ) – interacted with netrin-1, although with a reduced affinity (KdAβ/netrin–1:22 nM, KdAβ1–17/netrin-1: 30 nM; Figure 2d). Thus, the APP ectodomain interacts with netrin-1 at least in part in a region that includes the Aβ1–17 region of APP.
Netrin-1 affects APP signaling
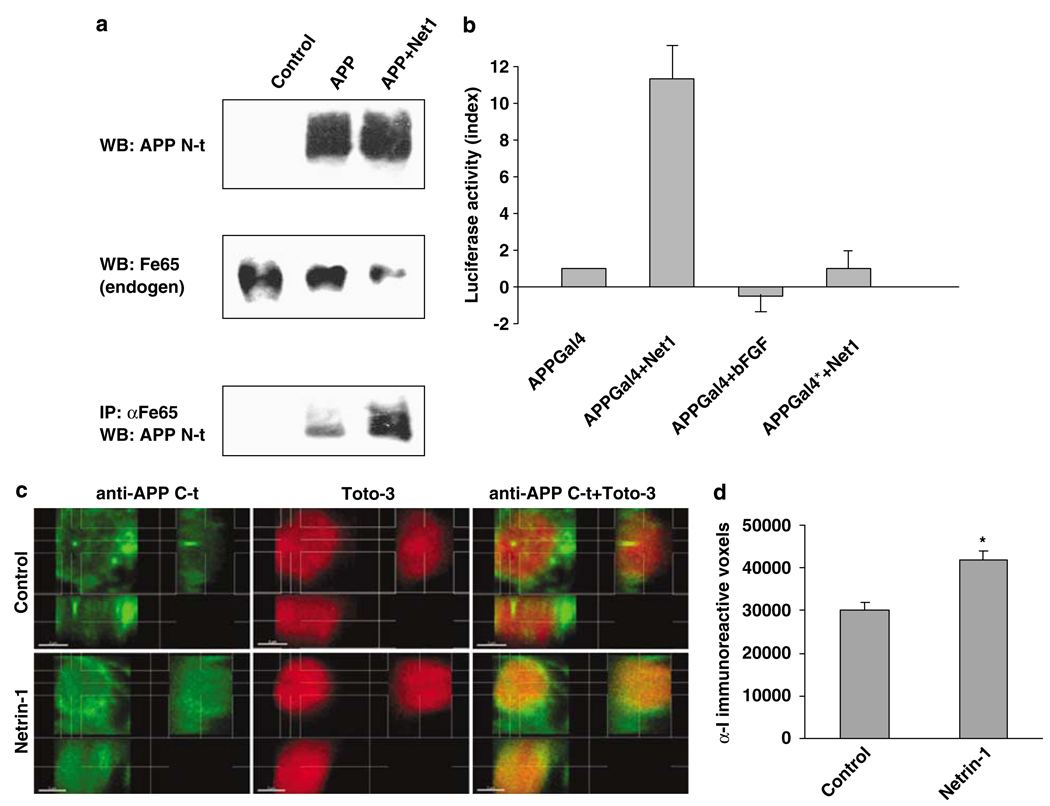
Figure 3 We next investigated whether netrin-1 may modulate the downstream signaling of APP. The adaptor protein DAB-1 has been shown to interact with the intracellular domain of APP. [15] As shown in Supplementary Figure 3a, netrin-1 enhanced the recruitment of DAB-1 to APP. Similarly, the adaptor protein Fe65 has been shown to interact with the intracellular domain of APP and to provide a mechanism for the coupling of APP to the cytoskeleton. [16] Fe65 has also been shown to be responsible for APP intracellular domain (AICD)-dependent gene transcription. Indeed, even though it is still a matter of debate, it is believed that in the presence of Fe65, the intracellular domain of APP migrates to the nucleus and initiates APP-dependent transcription of a specific set of genes through interaction with TIP60. [17–20] Figure 3a shows that, in APP-transfected B103 cells, the Fe65 interaction with APP is mainly observed when netrin-1 is added. According to the model proposed, this netrin-1-dependent enhanced interaction of Fe65 with APP could lead to an increased AICD activity. To test this, we first used the transactivation assay developed previously [17] in which the DNA-binding domain of Gal4 is fused to APP, and transactivation is monitored with a Gal4-dependent reporter plasmid.
As shown in Figure 3b, netrin-1 triggers APP-dependent gene transactivation in HEK293T, whereas bFGF has no effect on the Gal4-dependent reporter. This netrin-1-dependent, APP-dependent transactivation is abolished when APP is mutated at the Fe65 binding site. To further study netrin-1 effects on AICD, AICD was analyzed in primary cortical neurons derived from E16.5 embryos from transgenic mice expressing a human APP minigene carrying the Swedish and Indiana familial AD mutations (PDAPP). [21] As AICD-nuclear localization is an important – even though dispensable – event in AICD-dependent transactivation, [19] we assessed the effect of netrin-1 on the translocation of APP C-terminal-immunoreactive fragments into nuclei of hAPP transgenic cortical neurons. As shown in Figure 3c and d and Supplementary Figure 3bc, APP C-terminal-immunoreactive fragments are significantly increased in nuclei of netrin-1-treated neurons. We next determined whether KAI1, a gene whose expression has been shown to be AICD-dependent in different cell systems, [22] is induced in response to netrin-1. Therefore, quantitative RT-PCR on KAI1 was performed on RNA extracted from netrin-1-treated versus nontreated primary cortical neurons. Netrin-1 induced an increase in KAI1 expression (fold induction: 4.3±1.2). Thus, even though we cannot exclude the possibility that netrin-1-dependent KAI1 expression is due to a netrin-1 receptor other than APP, these observations, taken together with the transactivation assay performed in HEK293T cells, strongly support the notion that netrin-1 triggers AICD-dependent gene transactivation. Taken together, these results suggest that netrin-1 is likely to exert a functional effect on APP signaling.
Netrin-1 controls Aβ levels in an AD transgenic mouse model
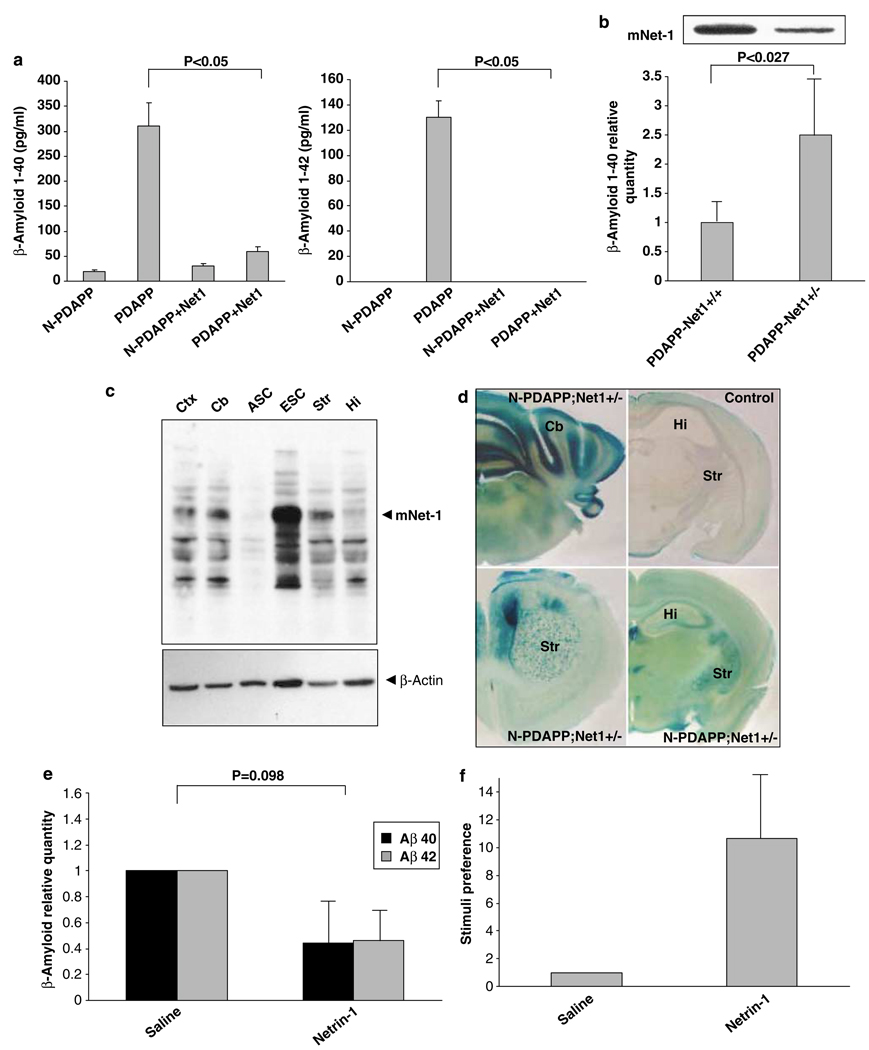
Figure 4 Because AD pathogenesis is thought to be mediated at least in part through APP processing resulting in Aβ production, we next evaluated the effect of netrin-1 on Aβ production in hAPP transgenic mice that model AD (PDAPP). [21] Whole brain slice cultures from PDAPP transgenic and nontransgenic littermates were treated with vehicle or with netrin-1 and were evaluated by ELISA assay for the production of Aβ1–40 and Aβ1–42. As shown in Figure 4a, the Alzheimer model transgenic mice displayed a marked increase in Aβ1–40 and Aβ1–42 net productions over that of the control mice (nontransgenic littermates), but this was suppressed by the addition of netrin-1. To assess the role of netrin-1 in a complementary, loss-of-function experiment, we crossed netrin-1 hemizygous mice (netrin nulls are nonviable) with hAPP transgenic mice, and then quantitated cerebral Aβ concentrations in the progeny by ELISA assay.
The PDAPP transgenic netrin-1 hemizygotes (PDAPP/netrin-1 +/–) showed a decreased netrin-1 level in the cortex compared to wild-type mice (PDAPP/netrin-1+/+; Figure 4b, inset). Quantitation of Aβ revealed a significant increase in Aβ levels in netrin-1 hemizygotes compared to wild-type mice (Figure 4b), hence strengthening the view of netrin-1 as a key regulator of Aβ level. Interestingly, netrin-1 is expressed not only during brain development but also in adult brain. Some brain regions still express high levels of netrin-1, as for example the striatum, the ventral tegmentum, and the substantia nigra (Figure 4c and d and Livesey and Hunt [23]). Although these brain regions are not those most commonly associated with pathology in AD, these regions have indeed been shown to be affected in Alzheimer’s. Furthermore, netrin-1 expression is also detected in the main regions of Aβ deposition and Alzheimer-related pathology including hippocampus and dentate gyrus (Figure 4c and d and Colicos [24]). It will, therefore, be of interest to determine whether the concentration ratio of Aβ peptides to netrin-1 in the adult brain represents a critical determinant of the development of AD.
Initial studies toward the use of netrin-1 or derivatives to ameliorate the Alzheimer’s phenotype
Netrin-1 thus shows multiple effects that make it a potential candidate for therapy in AD: (i) it markedly decreases Aβ1–40 and Aβ1–42 concentrations in brain slices from Alzheimer model transgenic mice; (ii) it interacts with Aβ peptide, with as-yet-unknown effects on Aβ oligomerization and clearance; (iii) it displays a neurotrophic effect. [25] As a first step toward a potential therapeutic use of netrin-1 in AD model transgenic mice, we delivered netrin-1 through the intracerebroventricular pump injection in PDAPP mice. As shown in Figure 4e, a 13-day netrin-1 brain injection (but not vehicle injection) was associated with a decreased Aβ level. Furthermore, consistent with the observed reduction in Aβ levels in netrin-1-treated Alzheimer’s mice, preliminary results suggest that PDAPP mice infused with netrin-1 showed significantly improved memory function. Indeed, as reported previously, [26, 27] PDAPP mice lacked the normal preference for novel stimuli, a trend that was exacerbated in the olfactory novelty (ON) recognition tests (Figure 4f).
In contrast, preliminary results show that netrin-1-infused PDAPP mice have a pronounced increase in their preference for the exploration of novel stimuli (Figure 4f). Even though we cannot exclude the possibility that this netrin-1-dependent memory enhancement was mediated by a receptor other than APP, this observation strengthens the rationale for mimicking netrin-1 as a putative treatment in AD. Thus, although further work will be required to prove that the delivery of netrin-1, a netrin-1 domain, or a netrin-1 mimetic may be of interest for the amelioration of AD pathology in this transgenic mouse model, the observations that netrin-1 acts as a ligand for APP, controls Aβ level, and may positively impact memory function, opens a novel avenue of research and potential application that links netrin-1, APP-mediated signal transduction, Aβ synthesis, neurite extension/retraction, and AD.
Materials and Methods
Cells, transfection procedures, and purified and recombinant proteins
Transfections of HEK293T (human embryonic kidney) or B103 (neuroblastoma) cells were performed using the lipofectamine reagent (Invitrogen, Carlsbad, CA, USA). Primary cortical neurons were obtained from E16.5 embryos and cultured in neurobasal media supplemented with B27 (Invitrogen). Recombinant Flag-netrin-1 was from Apotech Corp (Axxora). C-myc-tagged netrin-1 was purified from netrin-1-producing 293-EBNA cells as before. [8] GST/netrin-1 was produced as described previously. [28] His-netrin-1 was from R&D systems (Minneapolis, MN, USA). bFGF was from ABcam. Recombinant APPs, Aβ1–42 (and Aβ1–17), and DCC-EC-Fc were purchased from Sigma Aldrich, Anaspec, and R&D systems, respectively.
Plasmid constructs
Full-length APP695 (pcDNA3-APP), netrin-1 (pGNET1–myc), netrin-2 (pGNET2-myc), netrin G1 (pcDNA4-G1-netrin), domains V and VI of netrin-1 (pCEP4-netV-VI), APP-C100 (pcINeo-C100), APLP1 (pcINeo-APLP1), and DCC (pCMV-DCC-S)-expressing constructs were described previously. [8, 10, 29–32] APP613-695 was obtained by PCR using pcINeo-C100 as a PCR template and the following primers: 5'-CACCATGTTGGTGTTCTTTGCAGA-3' and 5'-CTAGTT CTGCATCTGCTCAAA-3', and inserted into a pcDNA3.1TOPOd (Invitrogen). PG5E1B-luc (Gal4 reported construct), pMst (Gal4), pMst-APP (APP-Gal4), pMst-APP* (mutated form of APP unable to bind to Fe65) were described previously. [17] TrkC (pCDNA3-TrkC-HA) was obtained by inserting into pCDNA3 vector, the rat TrkC coding sequence obtained from a pCMX-Trkc plasmid kindly provided by S Meakey.
APP/netrin-1 detection by ELISA assay
Here, 96-well plates (Immnunoplate Maxisorp, Nunc) were coated overnight at room temperature with a secreted form of APP (Aβ and Aβ1–17, respectively ) at a concentration of 2.5 µg/ml (respectively: 2.5, 0.18, 0.07 µg/ml). After 1 h blocking at 37°C with 5%FCS in PBS, wells were washed (0.05%Tween20 in PBS), followed by netrin-1 (Apotech) or bFGF incubation in a concentration range from 0.225 nM-60 nM at 37°C for 1 h. Washed wells were incubated with an anti-FlagM2 (Sigma-Aldrich) or anti-bFGF antibody in blocking buffer for 30 min at 37°C. An antimouse antibody coupled to HRP (Jackson ImmunoResearch Inc.) was added at a concentration of 0.8 µg/ml for 1 h at 37°C. Colorimetric intensity was measured at a wavelength of 490 nm using a Victor station (Wallac).
Western blot and immunoprecipitation
Western blots were performed as described, [12] using APP (C-terminal epitope:Sigma-Aldrich; N-terminal epitope: Sigma-Aldrich), Aβ (biosource), Flag M2 (Sigma-Aldrich), DAB-1 (Exalpha Biologicals), Fe65 (Upstate), GST (Sigma-Aldrich),c-myc (Sigma-Aldrich), P75-NGFR (abcam), TrkC (Santa-Cruz), or Net-1 antimouse monoclonal (R&D systems) antibody. Coimmunoprecipitations from HEK293 and B103 were performed as described previously. [33] Coimmunoprecipitations from primary culture were performed using the microbeads system developed by Miltenyi Biotech. Immunoprecipitation in developing brain was performed with a mixed mouse monoclonal anti-APP (5A3/ 1G7) antibody provided by E Koo. APP mutant mice were in a mixed background of C57BL6, 129SvEv, and 129 Ola and were described before. [34] E14.5 mice embryos originating from the same cross APP+/–xAPP+/– were further dissected while genotyping was performed a posteriori.
Confocal analysis on primary cultures of neurons
Primary cultures of neurons were fixed in 4% PFA and stained with various primary antibodies (5A3/1G7, antinetrin 64, or antiserum I (R1155)) followed by Alexa568- and/or Alexa488-conjugated antimouse and/or antirabbit secondary antibodies, respectively. Stacks of images (z-step=0.25 µm) were acquired with a laser scanning confocal microscope (Nikon PCM-2000) using a 100X objective and a 2.7 digital zoom, collected using SimplePCI software (Compix Inc., Lake Oswego, OR, USA) and processed in an SGI Octane R12 computer running Bitplane’s Advanced Imaging Software suite.
Organotypic culture and Aβ release determination
PDAPP(J20) mice were described before. [27] In all, 250 µm coronal brain slices were cut from P1 transgenic and nontransgenic littermates’ whole brain to establish organotypic cultures. Tissues were incubated in 0.5% D-glucose, 25% fetal bovine serum, 25% Hank’s buffered saline solution, and in Opti-MEM (Invitrogen). Recombinant netrin-1, NGF, or IGF-1 was added to the media immediately after plating and every 24 h. After 3 days, Aβ1–40 and Aβ1–42 were quantitated in the culture media using specific ELISA assays (Biosource, Camarillo, CA, USA). To measure Aβ in a context of lower netrin-1 concentration, transgenic PDAPP(J20) mice were crossed with netrin-1 +/– mice. Netrin-1 mutant mice were described before. [9, 35] Mice with adequate genotypes (PDAPP/netrin-1+/– and PDAPP/netrin-1 +/+) were analyzed for Aβ level using the specific above ELISA assay.
Intracerebroventricular pump injection and behavioral testing in PDAPP mice
Recombinant mouse netrin-1 (R&D) was infused in the left ventricle using Alzet osmotic pumps (Alzet 1002, 0.25 µl/h release rate) for 12–13 days. To assess memory, the simple ON test was used essentially as described [27] and in accordance with the ON SOP from the Murine Behavioral Assessment Laboratory at the University of California, Davis, CA, USA) 1 day before euthanasia. Preference for novelty in the ON test depends on the ability of mice to recall prior experience when presented with a previously encountered scent together with a novel one. Percentage of total time spent investigating the novel scent (preference) was scored and used as a measure of memory of previous experience.
Supplementary Material
Acknowledgements
We thank E Koo, V Castellani for discussion, H Arakawa for the GST/netrin-1-expressing construct and T Südhof for the APP-Gal4 transactivation system. We thank C Guix, MM Coissieux, A Tang, O Gorostiza and D Crippen for excellent technical assistance and C Ségura-Ferlay for statistical analysis. This work was supported by the Ligue Contre le Cancer (PM), the fondation pour le Cerveau (PM), the ARC (PM), the ANR (PM), the NIH (NS33376 to PM and DEB), the Joseph Drown Foundation (DEB), the John Douglas French Foundation (VG), and the Alzheimer’s Association (VG). VG thanks Mrs Eloise Goodhew Barnett for her support. FC is supported by a fellowship from the Portuguese Science and Technology Foundation (POCI2010).
Abbreviations
AD = Alzheimer’s disease
Aβ = amyloid-β
APP = amyloid precursor protein
DCC = deleted in colorectal cancer
APLP1 = amyloid precursor-like protein 1
FGF = fibroblast growth factor
Conflict of interest.
Authors declare to have no conflict of interest.
References:
Hansen LA, Terry RD.
Position paper on diagnostic criteria for Alzheimer disease.
Neurobiol Aging. 1997;18(4 Suppl):S71–S73Koo EH.
The beta-amyloid precursor protein (APP) and Alzheimer’s disease: does the tail wag the dog?
Traffic. 2002;3:763–770Turner PR, O'Connor K, Tate WP, Abraham WC.
Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory.
Prog Neurobiol. 2003;70:1–32Beher D, Hesse L, Masters CL, Multhaup G.
Regulation of amyloid protein precursor (APP) binding to collagen and mapping of the
binding sites on APP and collagen type I.
J Biol Chem. 1996;271:1613–1620Caceres J, Brandan E.
Interaction between Alzheimer’s disease beta A4 precursor protein (APP) and the extracellular matrix:
evidence for the participation of heparan sulfate proteoglycans.
J Cell Biochem. 1997;65:145–158Williamson TG, Mok SS, Henry A, Cappai R, Lander AD, Nurcombe V, et al.
Secreted glypican binds to the amyloid precursor protein of Alzheimer’s disease (APP)
and inhibits APP-induced neurite outgrowth.
J Biol Chem. 1996;271:31215–31221Ma QH, Futagawa T, Yang WL, Jiang XD, Zeng L, Takeda Y, et al.
A TAG1-APP signalling pathway through Fe65 negatively modulates neurogenesis.
Nat Cell Biol. 2008;10:283–294Serafini T, Kennedy TE, Galko MJ, Mirzayan C, Jessell TM, Tessier-Lavigne M.
The netrins define a family of axon outgrowth-promoting proteins homologous to C. elegans UNC-6.
Cell. 1994;78:409–424Serafini T, Colamarino SA, Leonardo ED, Wang H, Beddington R, Skarnes WC, et al.
Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system.
Cell. 1996;87:1001–1014Keino-Masu K, Masu M, Hinck L, Leonardo ED, Chan SS, Culotti JG, et al.
Deleted in colorectal cancer (DCC) encodes a netrin receptor.
Cell. 1996;87:175–185Mazelin L, Bernet A, Bonod-Bidaud C, Pays L, Arnaud S, Gespach C, et al.
Netrin-1 controls colorectal tumorigenesis by regulating apoptosis.
Nature. 2004;431:80–84Mehlen P, Rabizadeh S, Snipas SJ, Assa-Munt N, Salvesen GS, Bredesen DE.
The DCCgene product induces apoptosis by a mechanism requiring receptor proteolysis.
Nature. 1998;395:801–804Small DH, Nurcombe V, Reed G, Clarris H, Moir R, Beyreuther K, et al.
A heparin-binding domain in the amyloid protein precursor of Alzheimer’s disease is involved
in the regulation of neurite outgrowth.
J Neurosci. 1994;14:2117–2127Lu X, Le Noble F, Yuan L, Jiang Q, De Lafarge B, Sugiyama D, et al.
The netrin receptor UNC5B mediates guidance events controlling morphogenesis of the vascular system.
Nature. 2004;432:179–186Trommsdorff M, Borg JP, Margolis B, Herz J.
Interaction of cytosolic adaptor proteins with neuronal apolipoprotein E receptors and
the amyloid precursor protein.
J Biol Chem. 1998;273:33556–33560Ando K, Iijima KI, Elliott JI, Kirino Y, Suzuki T.
Phosphorylation-dependent regulation of the interaction of amyloid precursor protein
with Fe65 affects the production of beta-amyloid.
J Biol Chem. 2001;276:40353–40361Cao X, Sudhof TC.
A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and
histone acetyltransferase Tip60.
Science. 2001;293:115–120Kimberly WT, Zheng JB, Guenette SY, Selkoe DJ.
The intracellular domain of the beta-amyloid precursor protein is stabilized by Fe65 and
translocates to the nucleus in a notch-like manner.
J Biol Chem. 2001;276:40288–40292Cao X, Sudhof TC.
Dissection of amyloid-beta precursor protein-dependent transcriptional transactivation.
J Biol Chem. 2004;279:24601–24611Kimberly WT, Zheng JB, Town T, Flavell RA, Selkoe DJ.
Physiological regulation of the beta-amyloid precursor protein signaling domain by
c-Jun N-terminal kinase JNK3 during neuronal differentiation.
J Neurosci. 2005;25:5533–5543Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, et al.
High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice:
synaptotoxicity without plaque formation.
J Neurosci. 2000;20:4050–4058von Rotz RC, Kohli BM, Bosset J, Meier M, Suzuki T, Nitsch RM, et al.
The APP intracellular domain forms nuclear multiprotein complexes and regulates the
transcription of its own precursor.
J Cell Sci. 2004;117(Pt 19):4435–4448Livesey FJ, Hunt SP.
Netrin and netrin receptor expression in the embryonic mammalian nervous system suggests
roles in retinal, striatal, nigral, and cerebellar development.
Mol Cell Neurosci. 1997;8:417–429Colicos MA. thesis.
McGill University; 1999Llambi F, Causeret F, Bloch-Gallego E, Mehlen P.
Netrin-1 acts as a survival factor via its receptors UNC5 H and DCC.
EMBO J. 2001;20:2715–2722Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W, et al.
Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer
amyloid precursor proteins.
Neuron. 1995;15:1203–1218Galvan V, Gorostiza OF, Banwait S, Ataie M, Logvinova AV, Sitaraman S, et al.
Reversal of Alzheimer’s-like pathology and behavior in human APP transgenic mice by
mutation of Asp664.
Proc Natl Acad Sci USA. 2006;103:7130–7135Tanikawa C, Matsuda K, Fukuda S, Nakamura Y, Arakawa H.
p53RDL1 regulates p53-dependent apoptosis.
Nat Cell Biol. 2003;5:216–223Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, et al.
A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor.
Nat Med. 2000;6:397–404Lu DC, Shaked GM, Masliah E, Bredesen DE, Koo EH.
Amyloid beta protein toxicity mediated by the formation of amyloid-beta protein
precursor complexes.
Ann Neurol. 2003;54:781–789Galvan V, Chen S, Lu D, Logvinova A, Goldsmith P, Koo EH, et al.
Caspase cleavage of members of the amyloid precursor family of proteins.
J Neurochem. 2002;82:283–294Homayouni R, Rice DS, Sheldon M, Curran T.
Disabled-1 binds to the cytoplasmic domain of amyloid precursor-like protein 1.
J Neurosci. 1999;19:7507–7515Forcet C, Ye X, Granger L, Corset V, Shin H, Bredesen DE, et al.
The dependence receptor DCC (deleted in colorectal cancer) defines an alternative
mechanism for caspase activation.
Proc Natl Acad Sci USA. 2001;98:3416–3421Heber S, Herms J, Gajic V, Hainfellner J, Aguzzi A, Rulicke T, et al.
Mice with combined gene knock-outs reveal essential and partially redundant functions of
amyloid precursor protein family members.
J Neurosci. 2000;20:7951–7963Forcet C, Stein E, Pays L, Corset V, Llambi F, Tessier-Lavigne M, et al.
Netrin-1-mediated axon outgrowth requires deleted in colorectal cancer-dependent MAPK activation.
Nature. 2002;417:443–447Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S.
Automatic and quantitative measurement of protein–protein colocalization in live cells.
Biophys J. 2004;86:3993–4003Selkoe DJ, Podlisny MB, Joachim CL, Vickers EA, Lee G, Fritz LC, et al.
Beta-amyloid precursor protein of Alzheimer disease occurs as 110- to 135-kilodalton
membrane-associated proteins in neural and nonneural tissues.
Proc Natl Acad Sci USA. 1988;85:7341–7345.
Return to ALZHEIMER's
Since 4-10-2020
| Home Page | Visit Our Sponsors | Become a Sponsor |
Please read our DISCLAIMER |