

Preventing Alzheimer's Disease-related
Gray Matter Atrophy by B-vitamin TreatmentThis section is compiled by Frank M. Painter, D.C.
Send all comments or additions to: Frankp@chiro.org


FROM: Proc Natl Acad Sci U S A. 2013 (Jun 4); 110 (23): 9523–9528 ~ FULL TEXT
Douaud G, Refsum H, de Jager CA, Jacoby R, E Nichols T, Smith SM, Smith AD
Functional Magnetic Resonance Imaging of the Brain (FMRIB) Centre,
Nuffield Department of Clinical Neurosciences,
University of Oxford,
John Radcliffe Hospital,
Oxford OX3 9DU, United Kingdom.
Alzheimer's Changes Slowed with B Vitamins
A combination of B vitamins was found to slow brain loss in areas of the brain associated with Alzheimer's disease. This same combination of vitamins has also been shown previously to slow cognitive decline.
Don't waste your money at Walgreens. Take a high quality, all natural B Complex supplement that will actually deliver results.
Is it possible to prevent atrophy of key brain regions related to cognitive decline and Alzheimer's disease (AD)? One approach is to modify nongenetic risk factors, for instance by lowering elevated plasma homocysteine using B vitamins. In an initial, randomized controlled study on elderly subjects with increased dementia risk (mild cognitive impairment according to 2004 Petersen criteria), we showed that high-dose B-vitamin treatment (folic acid 0.8 mg, vitamin B6 20 mg, vitamin B12 0.5 mg) slowed shrinkage of the whole brain volume over 2 y. Here, we go further by demonstrating that B-vitamin treatment reduces, by as much as seven fold, the cerebral atrophy in those gray matter (GM) regions specifically vulnerable to the AD process, including the medial temporal lobe. In the placebo group, higher homocysteine levels at baseline are associated with faster GM atrophy, but this deleterious effect is largely prevented by B-vitamin treatment. We additionally show that the beneficial effect of B vitamins is confined to participants with high homocysteine (above the median, 11 µmol/L) and that, in these participants, a causal Bayesian network analysis indicates the following chain of events: B vitamins lower homocysteine, which directly leads to a decrease in GM atrophy, thereby slowing cognitive decline. Our results show that B-vitamin supplementation can slow the atrophy of specific brain regions that are a key component of the AD process and that are associated with cognitive decline. Further B-vitamin supplementation trials focusing on elderly subjets with high homocysteine levels are warranted to see if progression to dementia can be prevented.
Keywords: structural neuroimaging, hippocampus, causal modeling, degeneration, clinical trial
From the FULL TEXT Article:
Introduction
The prevention of Alzheimer’s disease (AD) is a major public health challenge, but several promising therapies targeting β-amyloid have failed in late-stage clinical trials [1]. An alternative approach is to modify nongenetic risk factors and to treat people at risk of developing dementia before they develop the major symptoms [2, 3]. Many cross-sectional and prospective studies have shown that raised levels of plasma total homocysteine (tHcy) are associated with cognitive impairment, AD, or vascular dementia [4–9], but randomized, controlled trials of tHcy-lowering treatment using B-vitamin supplementation have shown inconsistent results on cognitive function [10, 11]. Factors such as dosage, vitamin combination, duration of treatment and the population treated possibly account for some of the discrepancies [10, 11]. Another factor that may explain the discrepancies in these trials is baseline tHcy concentration: for instance, it has been found that subjects with modestly raised tHcy experience a beneficial effect of B-vitamin treatment on cognitive decline [12, 13].
For trials with the aim of slowing progression of cognitive decline, the usual design includes neuropsychological assessments. Unfortunately, such testing is subject to short-term fluctuations, practice effects and intra-/interrater variability. In contrast, structural neuroimaging provides a robust way of assessing changes of a longer-term nature, including the impact of the treatment [14–16]. Accordingly, we recently showed, in a first study on the same subjects as included here, that B-vitamin treatment slowed the shrinkage of the whole brain over 2 y and that there was an interaction between treatment and tHcy at baseline [17].
Since the measure of overall brain size used in the initial study was nonspecific, key questions remain to be elucidated:(i) are B vitamins effective in preventing the atrophy of those gray matter (GM) regions specifically affected by the AD neurodegenerative process,
(ii) does this effect differ according to baseline tHcy,
(iii) does the reduction of atrophy with B vitamins occur in regions linked to cognitive and clinical outcomes, and
(iv) can GM atrophy provide a biological intermediary in determining the causal chain of events linking B-vitamin treatment to changes in cognition?

Table 1 To answer these questions, we used the data from a randomized, controlled trial (VITACOG) assessing the effect of B-vitamin treatment (folic acid 0.8 mg/d, vitamin B12 0.5 mg/d, vitamin B6 20 mg/d) on elderly volunteers with memory complaint who fulfilled the Petersen criteria for mild cognitive impairment (MCI) [18] over a period of 24 mo. MRI scans at baseline and the second time point from 156 subjects, 76 receiving placebo and 80 receiving B vitamins (mean age: 76 and 77 y, respectively; Table 1), were suitable for analysis. Local GM volume at baseline and after 24 mo was assessed with the optimized voxel-based morphometry (VBM) protocol using FSL (FMRIB Software Library) [19–21]. Placebo and B-vitamin groups did not differ in baseline GM volume [minimum familywise error (FWE), P = 0.33; SI Methods].
Results
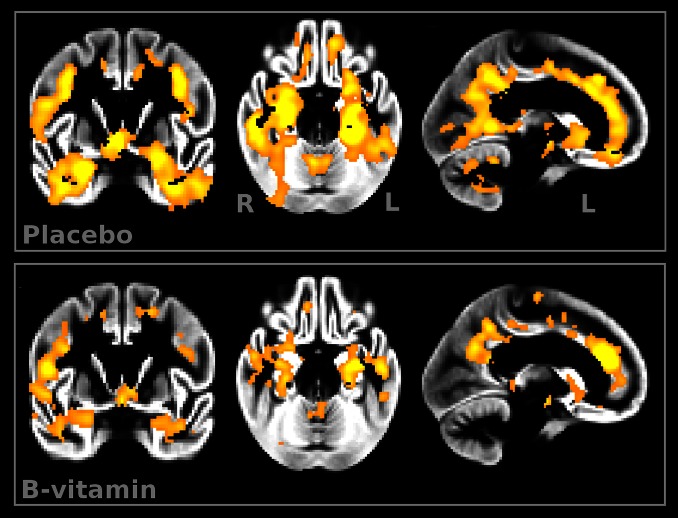
Figure 1
Table 2
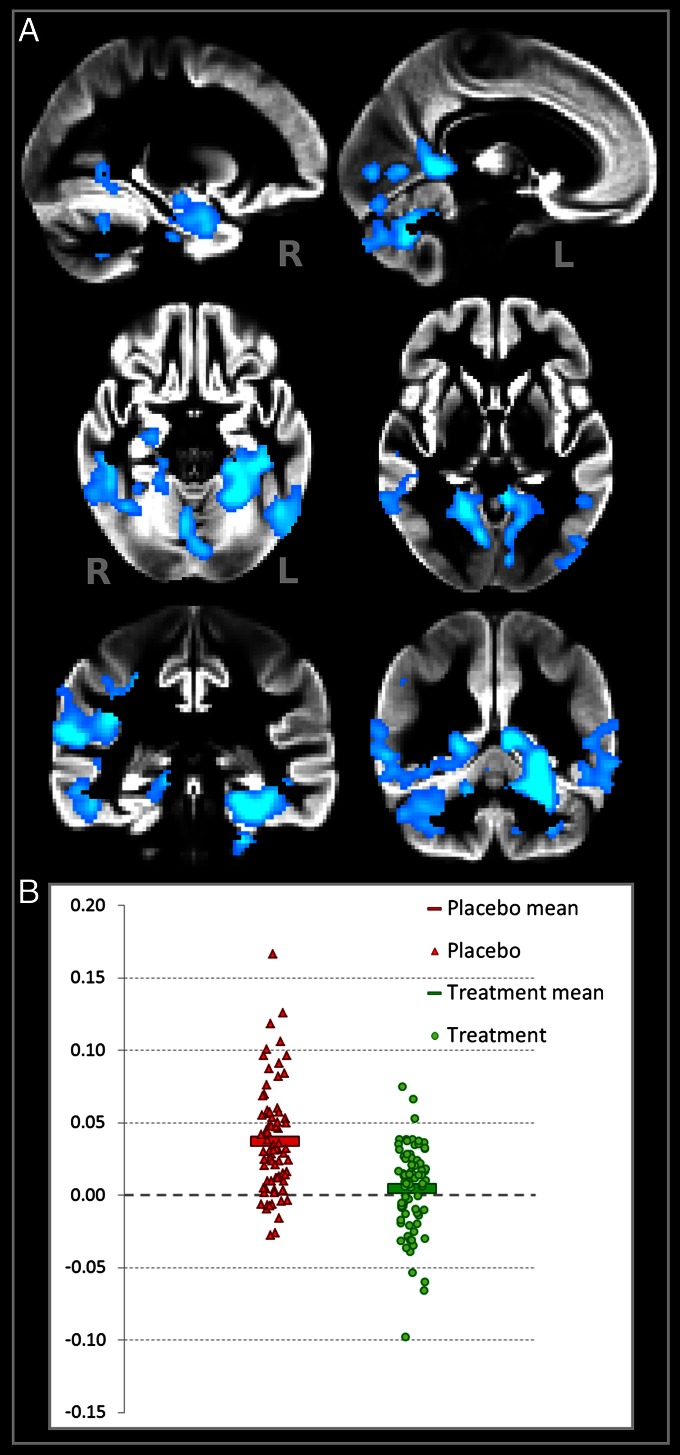
Figure 2
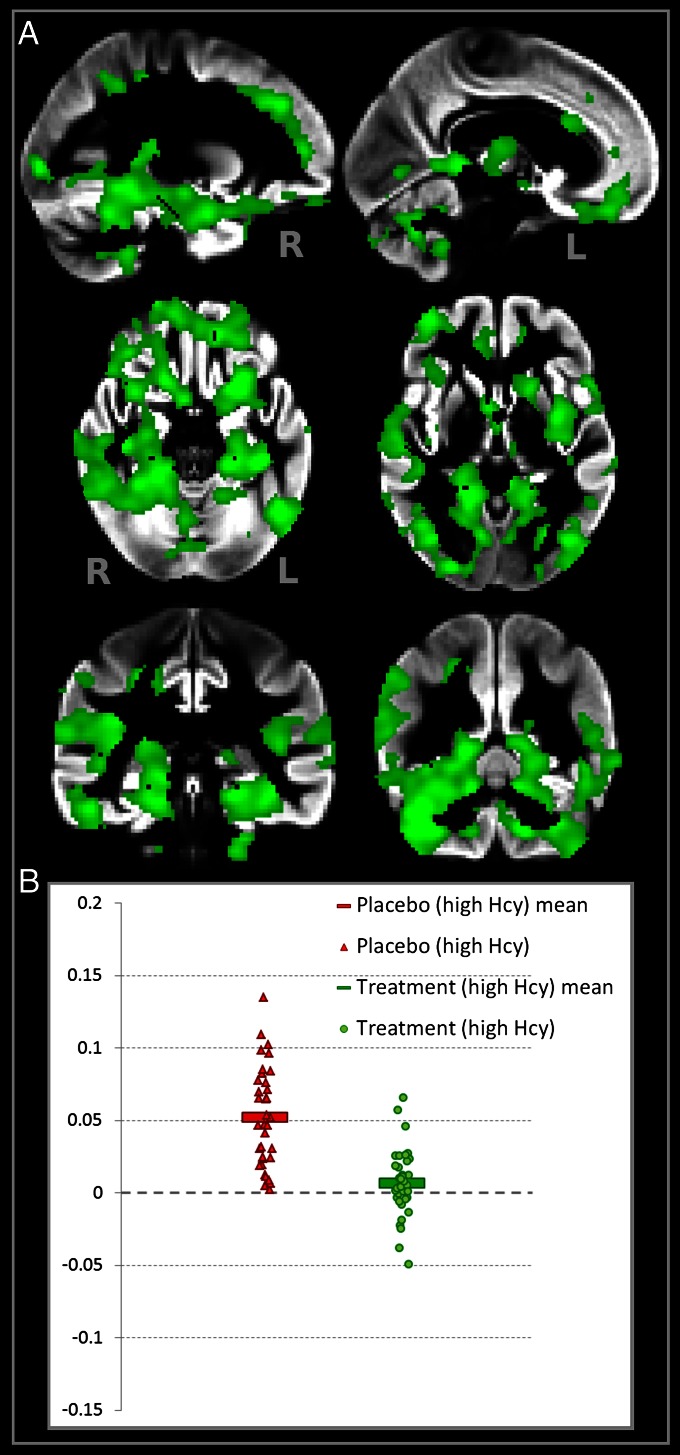
Figure 3
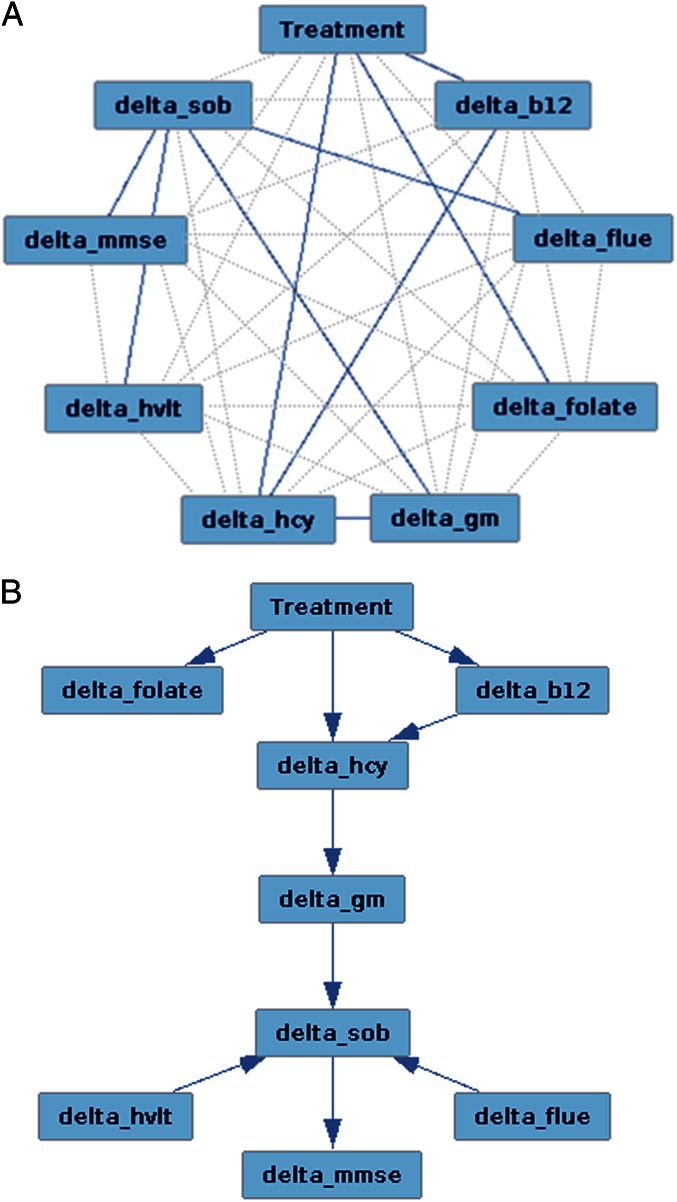
Figure 4 We first sought to establish whether the two treatment groups lost GM over the 2-y period. Atrophy was found in both placebo and B-vitamin groups in similar areas encompassing medial temporal, lateral temporoparietal, and occipital regions, as well as the anterior and posterior cingulate cortex (Figure 1). Direct comparison between groups revealed a significant effect of treatment: subjects receiving B vitamins showed a significant reduction of atrophy compared with the placebo group in posterior brain regions including bilateral hippocampus and parahippocampal gyrus, retrosplenial precuneus, lingual and fusiform gyrus, as well as in the cerebellum (Table 2). These regions are among those most affected in AD, and also in MCI subjects who later convert to AD [14, 15, 22–24]. In the regions showing significant treatment effect, the average loss of GM over 2 y was 3.7% (±3.7) in the placebo group compared with only 0.5% (±2.9) in the B-vitamin group (Figure 2 and Figure S1).
As previously shown [4, 17, 25–27], higher plasma tHcy levels are related to smaller global brain volume and white matter volume, smaller amygdala and hippocampus, and faster reduction in overall brain size. Participants were divided into two groups according to baseline tHcy: those with measurements below and above the median (11.06 µmol/L). We investigated the interplay between treatment and baseline tHcy. First, we tested the effect of baseline tHcy on atrophy in the placebo and B-vitamin groups separately. Participants with high tHcy in the placebo group had greater GM atrophy compared with those with low tHcy (Fig. S2). In contrast, in the subjects receiving B vitamins, there was no difference in atrophy between participants with low and high baseline tHcy, even when looking at the entire distribution of t values over the brain [median t(placebo) = 0.53; median t(B vitamin) = -0.04]. Second, we tested the effect of treatment on atrophy in subjects with high and low tHcy levels separately. No significant effect of B-vitamin treatment was found in the participants with low baseline tHcy (minimum FWE, P = 0.35). However, B-vitamin treatment had a marked beneficial effect in reducing GM atrophy over 2 y in those with high tHcy [from 5.2% (±3.4) down to 0.6% (±2.1); Fig. 3]. In the high tHcy group, B vitamins reduced atrophy in similar regions as seen in the total group, and it also extended to anterior regions including the anterior cingulate cortex and piriform cortex, as well as prefrontal areas (Figure 3, Fig. S3, and Table 2). A formal interaction between treatment status and baseline tHcy also proved significant (Fig. S4).
To establish which regions of the brain were associated with cognitive decline, we performed voxelwise linear regression analyses between change in GM volume and change in neuropsychological scores over time on all 156 participants. More precisely, we explored the relationship between GM loss and decline in measures of global function (CDR-SOB and MMSE), decline in episodic memory (HVLT-R), and decline in semantic memory (category fluency). Regression analyses revealed significant association of GM loss with worsening of CDR-SOB and MMSE scores, which was most pronounced bilaterally in the amygdalohippocampal complex and entorhinal cortex. At an uncorrected threshold (voxelwise P < 0.01), decreases in HVLT-R delayed recall and category fluency were associated with increased GM loss in the left hippocampus and entorhinal cortex (Fig. S5). These GM regions involved in cognitive decline also showed a reduction of atrophy with B-vitamin treatment in subjects with high tHcy levels (Fig. S6).
We finally sought to determine the role of tHcy and brain atrophy in mediating the influence of B-vitamin treatment on cognitive changes in participants with higher tHcy levels, as those were the only subjects to benefit from the treatment. We modeled treatment and change in plasma concentrations of tHcy and relevant B vitamins, imaging, and neuropsychological measures over the 2-y period as a directed acyclic graph. We found that the optimal Bayesian network explaining our data suggested the following chain of events: treatment led to a change in vitamin B12 and folate plasma concentrations, with only vitamin B12 appearing to play a role in modifying tHcy levels; changes in tHcy levels caused a change in GM atrophy, which, in turn, led to a modification of the CDR-SOB (Figure 4).
Discussion
Here, in an unbiased, voxel-based analysis, we have demonstrated that B-vitamin treatment, by lowering mean plasma tHcy levels (by 29%; SI Methods), markedly reduces GM atrophy in regions particularly susceptible to AD. The loss of total GM volume over the 2-y period (2.0% in the placebo group, 1.4% in the B-vitamin group; SI Methods) was comparable to yearly measures of the overall brain atrophy rate obtained in our previous study (1.08% and 0.76%, respectively) [17]. The regions significantly benefiting from the treatment thus show less atrophy relative to the total GM volume loss in the B-vitamin group (0.5% vs. 1.4%), whereas the same regions in the placebo group show an accelerated atrophy compared with the whole GM (3.7% vs. 2.0%), suggesting this set of regions is highly vulnerable to the possible underlying pathological process. Some of the GM regions that benefit the most from the B-vitamin treatment (hippocampus, parahippocampal gyrus, inferior parietal lobule and retrosplenial cortex) are all part of a “parieto–medial temporal pathway” involved in visuospatial learning and spatial long-term memory [28]. Notably, these regions, together with the fusiform and inferior temporal gyrus, which overall show the most significant reduction of atrophy by B vitamins in participants with high tHcy levels, are also among the best to discriminate between healthy aging and MCI, and all show the greatest atrophy rate in the progression from MCI to AD [23, 29, 30]. We also found some significant differences within the cerebellum. This result might seem surprising at first, but there is now clear anatomical and functional evidence for the role of the cerebellum in cognitive and behavioral functions, and particularly for autobiographical memory and working memory [31, 32]. Accordingly, our strongest results in the cerebellum are localized to regions that are known to be connected to the medial temporal lobe and prefrontal cortex [31, 33]. As part of the default mode network and of the network defined by an encoding memory paradigm, these cerebellar regions have also been shown to be altered in healthy subjects with an increased risk for dementia [34].
When splitting the groups according to the overall baseline tHcy median value, we further showed that the treatment was significantly beneficial only to participants with higher levels of tHcy, in line with the cognitive outcomes shown in this population [13]. In these participants with high tHcy levels, there was again a clear difference in loss of GM between regions significantly benefiting from vitamin B and the entire GM: less atrophy in the B-vitamin group (0.6% vs. 1.6%), accelerated atrophy in the placebo group (5.2% vs. 2.7%). In the B-vitamin group, we found no GM atrophy difference between those with low and high baseline tHcy, suggesting that taking B vitamins mimics the protective effect over time of having lower baseline tHcy. While vitamin B12 deficiency may lead to neurological and neuropsychiatric disorders, no participant had vitamin B12 deficiency at baseline in this study (<150 pmol/L). Our network analysis also suggests that changes in GM atrophy, while significantly related to a change in vitamin B12 levels, appear to be mediated via lowering of tHcy levels. The apparent lack of influence of changes in folate levels may be because, in populations with good folate status as here (mean: 29 nmol/L), the main determinant of tHcy levels is vitamin B12 status [35].
A plausible hypothesis to explain why lowering tHcy might decrease the rate of atrophy of the specific brain regions involved in AD is that the atrophy of such regions is related to the presence of neurofibrillary tangles [36]. The deposition of neurofibrillary tangles is caused by the formation of phosphorylated τ (tau), and raised tHcy concentrations lead to an increase in phosphorylated τ (tau) [37, 38]. Thus, lowering tHcy may decrease levels of phosphorylated τ (tau) and thereby reduce the degree of regional GM atrophy. Another possible explanation might stem from the converging evidence of an impaired neuroregeneration seen in AD [39, 40]. In the adult, neurogenesis occurs in the hippocampus and olfactory bulb, whereas evidence for it taking place in regions such as the prefrontal, posterior parietal, and inferior temporal cortex, as well as piriform cortex and amygdala, remain controversial [41]. Epigenetic mechanisms, involving DNA and histone methylation, are important for neurogenesis [42]. Raised tHcy is an inhibitor of methylation reactions and can inhibit the proliferation of neuronal cell precursors [43, 44]. As one of the main brain functions of vitamin B12 is in methylation [43] and as B12 deficiency can also impair neuronal cell proliferation [45], it is thus conceivable that vitamin B12 helps to maintain adult neurogenesis in the very regions targeted by AD.
By using measures of GM structure to robustly assess the effect of B-vitamin supplementation and by characterizing the relationship between atrophy in specific GM regions and cognitive decline, we have been able to demonstrate the benefit of this treatment in those elderly subjects with high levels of tHcy. High-dose B-vitamin treatment could modify a key component of the disease process leading to AD: the atrophy of GM regions involved in the cognitive decline of the study participants. Importantly, we also show that B-vitamin treatment is of no benefit for those participants with low levels of tHcy. While our voxel-wise, whole GM analysis was used to map the possible effect of treatment in an unbiased manner, it is also a limitation of this study in the context of a randomized controlled trial. First, such a voxelwise approach, as it encompasses data from ~200,000 voxels, is potentially more susceptible to Type I error compared with a region-of-interest approach, which is the method usually favored for clinical trials. Another limitation is that the results described here are parts of the secondary outcomes of the clinical trial as defined in our prespecified plan of data analysis (more specifically, of a regional brain volume changes analysis), while the primary endpoint of this trial was the impact of B vitamins on overall brain atrophy [17]. We note, however, that the statistical analyses carried out in this study and presented in Figs. 1–3 were all determined a priori and were performed blind to the treatment group. Baseline comparisons, effect of ApoE ε4 on GM atrophy and causal Bayesian network analysis (Fig. 4) were carried out a posteriori. Finally, while our voxel-based analyses must be strictly regarded as post hoc in the context of the clinical trial, they were fully corrected for multiple comparisons with stringent control of Type I error (i.e. with the Family Wise Error rate, instead of, e.g., False Discovery Rate) using nonparametric resampling-based inference that makes only weak assumptions on the data; thus, they comply with best practice for confirmatory inference in neuroimaging [46].
MCI is common among elderly populations, with approximately 16% of those older than 70 y of age showing this syndrome [47]. In population studies, it has been found that plasma tHcy concentrations increase with age. The proportion with concentrations >13 µmol/L, even in countries with mandatory folic acid fortification, ranges from 9% to 14% of those older than 60 y [48]. Thus, a significant proportion of elderly subjects may be at risk for dementia as a result of elevated tHcy (a Population-Attributable Risk of 16% has, for instance, been estimated in the Framingham Study) [6]. Larger and longer trials will be needed(i) to determine the optimal tHcy threshold warranting B-vitamin supplementation and
(ii) to monitor the treatment effect on the crucial outcome, the incidence of dementia.
Methods
This study was approved by the Oxfordshire National Health Service research ethics committee A on January 6, 2006 (Central Office for Research Ethics Committees, COREC 04/Q1604/100). 156 volunteers who fulfilled the Petersen criteria [18] for MCI underwent the same longitudinal imaging protocol with structural, high-resolution T1-weighted images acquired at baseline and after 2 y on a 1.5T Sonata MRI system (1-mm3 isotropic resolution; Siemens Medical Systems). An optimized FSL-VBM analysis was carried out (fsl.fmrib.ox.ac.uk/fsl/fslwiki/FSLVBM) [20, 21], with all images from both time points processed following the same protocol to avoid the known bias of using one of the two time points as reference. To achieve accurate inference, including FWE correction for multiple comparisons across space, we used permutation-based nonparametric inference within the framework of the general linear model (5,000 permutations) [49]. We tested for GM changes between baseline and follow-up scan in placebo and B-vitamin groups, and for differences in GM changes between the two populations. The B-vitamin and placebo groups were further divided according to their tHcy levels at baseline (≤11.06 µmol/L or >11.06 µmol/L, the median value). Results were considered significant at P < 0.05, FWE-corrected for multiple comparisons across space using Threshold-Free Cluster Enhancement [50]. All other statistical tests were carried out in R. Network analysis [51] was performed using Tetrad (www.phil.cmu.edu/projects/tetrad/). Goodness of fit was determined with a χ2 test. Using a subsampling procedure, we further verified the stability of the optimized network. More details of the method are provided in SI Methods.
Acknowledgments
The authors thank all participants in the trial and the entire OPTIMA team, Dr. Joe Ramsey for advice on the use of Tetrad, and Dr. Rita Marasco for help on the visual inspection of the scans. This work was supported by grants from the Charles Wolfson Charitable Trust, Medical Research Council, Alzheimer’s Research Trust, Henry Smith Charity, John Coates Charitable Trust, Thames Valley Dementias and Neurodegenerative Diseases Research Network of the National Institute for Health Research (United Kingdom), Sidney and Elizabeth Corob Charitable Trust and Meda AB/Recip (Solna, Sweden). Meda AB/Recip supplied the vitamin and placebo tablets.
References
Miller G.
Alzheimer’s research. Stopping Alzheimer’s before it starts.
Science. 2012;337(6096):790–792Sperling RA, Jack CR, Jr, Aisen PS.
Testing the right target and right drug at the right stage.
Sci Transl Med. 2011;3(111):111cm33Barnes DE, Yaffe K.
The projected effect of risk factor reduction on Alzheimer’s disease prevalence.
Lancet Neurol. 2011;10(9):819–828Clarke R, et al.
Folate, vitamin B12, and serum total homocysteine levels in confirmed Alzheimer disease.
Arch Neurol. 1998;55(11):1449–1455McCaddon A, Davies G, Hudson P, Tandy S, Cattell H.
Total serum homocysteine in senile dementia of Alzheimer type.
Int J Geriatr Psychiatry. 1998;13(4):235–239Seshadri S, et al.
Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease.
N Engl J Med. 2002;346(7):476–483Dufouil C, Alpérovitch A, Ducros V, Tzourio C.
Homocysteine, white matter hyperintensities, and cognition in healthy elderly people.
Ann Neurol. 2003;53(2):214–221Smith AD.
The worldwide challenge of the dementias: A role for B vitamins and homocysteine?
Food Nutr Bull. 2008;29(2, suppl):S143–S172Wald DS, Kasturiratne A, Simmonds M.
Serum homocysteine and dementia: Meta-analysis of eight cohort studies including 8669 participants.
Alzheimers Dement. 2011;7(4):412–417Wald DS, Kasturiratne A, Simmonds M.
Effect of folic acid, with or without other B vitamins, on cognitive decline: Meta-analysis of randomized trials.
Am J Med. 2010;123(6):522–527Ford AH, Almeida OP.
Effect of homocysteine lowering treatment on cognitive function: A systematic review and meta-analysis of randomized controlled trials.
J Alzheimers Dis. 2012;29(1):133–149Durga J, et al.
Effect of 3-year folic acid supplementation on cognitive function in older adults in the FACIT trial: A randomised, double blind, controlled trial.
Lancet. 2007;369(9557):208–216de Jager CA, Oulhaj A, Jacoby R, Refsum H, Smith AD.
Cognitive and clinical outcomes of homocysteine-lowering B-vitamin treatment in mild cognitive impairment: A randomized controlled trial.
Int J Geriatr Psychiatry. 2012;27(6):592–600Fox NC, et al.
Imaging of onset and progression of Alzheimer’s disease with voxel-compression mapping of serial magnetic resonance images.
Lancet. 2001;358(9277):201–205Scahill RI, Schott JM, Stevens JM, Rossor MN, Fox NC.
Mapping the evolution of regional atrophy in Alzheimer’s disease: Unbiased analysis of fluid-registered serial MRI.
Proc Natl Acad Sci USA. 2002;99(7):4703–4707Jack CR, Jr, et al.
Comparison of different MRI brain atrophy rate measures with clinical disease progression in AD.
Neurology. 2004;62(4):591–600Smith AD, et al.
Homocysteine-lowering by B Vitamins Slows the Rate of Accelerated Brain Atrophy in Mild Cognitive Impairment: A Randomized Controlled Trial
PLoS One. 2010 (Sep 8); 5 (9): e12244Petersen RC.
Mild cognitive impairment as a diagnostic entity.
J Intern Med. 2004;256(3):183–194Good CD, et al.
A voxel-based morphometric study of ageing in 465 normal adult human brains.
Neuroimage. 2001;14(1 pt 1):21–36Douaud G, et al.
Anatomically related grey and white matter abnormalities in adolescent-onset schizophrenia.
Brain. 2007;130(pt 9):2375–2386Smith SM, et al.
Advances in functional and structural MR image analysis and implementation as FSL.
Neuroimage. 2004;23(suppl 1):S208–S219Fennema-Notestine C, et al.
Alzheimer’s Disease Neuroimaging Initiative Structural MRI biomarkers for preclinical and mild Alzheimer’s disease.
Hum Brain Mapp. 2009;30(10):3238–3253Ferreira LK, Diniz BS, Forlenza OV, Busatto GF, Zanetti MV.
Neurostructural predictors of Alzheimer’s disease: A meta-analysis of VBM studies.
Neurobiol Aging. 2011;32(10):1733–1741Douaud G, et al.
Brain microstructure reveals early abnormalities more than two years prior to clinical progression from mild cognitive impairment to Alzheimer’s disease.
J Neurosci. 2013;33(5):2147–2155den Heijer T, et al.
Hippocampal, amygdalar, and global brain atrophy in different apolipoprotein E genotypes.
Neurology. 2002;59(5):746–748Seshadri S, et al.
Association of plasma total homocysteine levels with subclinical brain injury: Cerebral volumes, white matter hyperintensity, and silent brain infarcts at volumetric magnetic resonance imaging in the Framingham Offspring Study.
Arch Neurol. 2008;65(5):642–649Rajagopalan P, et al.
Homocysteine effects on brain volumes mapped in 732 elderly individuals.
Neuroreport. 2011;22(8):391–395.Kravitz DJ, Saleem KS, Baker CI, Mishkin M.
A new neural framework for visuospatial processing.
Nat Rev Neurosci. 2011;12(4):217–230Desikan RS, et al.
Automated MRI measures identify individuals with mild cognitive impairment and Alzheimer’s disease.
Brain. 2009;132(pt 8):2048–2057Desikan RS, et al.
MRI measures of temporoparietal regions show differential rates of atrophy during prodromal AD.
Neurology. 2008;71(11):819–825Schmahmann JD.
The role of the cerebellum in cognition and emotion: Personal reflections since 1982 on the dysmetria of thought hypothesis, and its historical evolution from theory to therapy.
Neuropsychol Rev. 2010;20(3):236–260Svoboda E, McKinnon MC, Levine B.
The functional neuroanatomy of autobiographical memory: A meta-analysis.
Neuropsychologia. 2006;44(12):2189–2208Habas C, et al.
Distinct cerebellar contributions to intrinsic connectivity networks.
J Neurosci. 2009;29(26):8586–8594Filippini N, et al.
Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele.
Proc Natl Acad Sci USA. 2009;106(17):7209–7214Green R, Miller JW.
Vitamin B12 deficiency is the dominant nutritional cause of hyperhomocysteinemia in a folic acid-fortified population.
Clin Chem Lab Med. 2005;43(10):1048–1051Whitwell JL, et al.
MRI correlates of neurofibrillary tangle pathology at autopsy: A voxel-based morphometry study.
Neurology. 2008;71(10):743–749Popp J, et al.
Homocysteine metabolism and cerebrospinal fluid markers for Alzheimer's disease.
Journal of Alzheimer's disease: JAD. 2009;18:819–828Zhang CE, et al.
Homocysteine induces tau phosphorylation by inactivating protein phosphatase 2A in rat hippocampus.
Neurobiol Aging. 2008;29(11):1654–1665Zhao C, Deng W, Gage FH.
Mechanisms and functional implications of adult neurogenesis.
Cell. 2008;132(4):645–660Mu Y, Gage FH.
Adult hippocampal neurogenesis and its role in Alzheimer's disease.
Molecular neurodegeneration. 2011;6:85Gould E.
How widespread is adult neurogenesis in mammals?
Nat Rev Neurosci. 2007;8(6):481–488Ma DK, et al.
Epigenetic choreographers of neurogenesis in the adult mammalian brain.
Nat Neurosci. 2010;13(11):1338–1344Weir DG, Scott JM.
Brain function in the elderly: Role of vitamin B12 and folate.
Br Med Bull. 1999;55(3):669–682Rabaneda LG, et al.
Homocysteine inhibits proliferation of neuronal precursors in the mouse adult brain by impairing the basic fibroblast growth factor signaling cascade and reducing extracellular regulated kinase 1/2-dependent cyclin E expression.
FASEB J. 2008;22(11):3823–3835Battaglia-Hsu SF, et al.
Vitamin B12 deficiency reduces proliferation and promotes differentiation of neuroblastoma cells and up-regulates PP2A, proNGF, and TACE.
Proc Natl Acad Sci USA. 2009;106(51):21930–21935Carter CS, Heckers S, Nichols T, Pine DS, Strother S.
Optimizing the design and analysis of clinical functional magnetic resonance imaging research studies.
Biol Psychiatry. 2008;64(10):842–849Petersen RC, et al.
Mild cognitive impairment: Ten years later.
Arch Neurol. 2009;66(12):1447–1455Pfeiffer CM, et al.
Trends in circulating concentrations of total homocysteine among US adolescents and adults: Findings from the 1991-1994 and 1999-2004 National Health and Nutrition Examination Surveys.
Clin Chem. 2008;54(5):801–813Nichols TE, Holmes AP.
Nonparametric permutation tests for functional neuroimaging: A primer with examples.
Hum Brain Mapp. 2002;15(1):1–25Smith SM, Nichols TE.
Threshold-free cluster enhancement: Addressing problems of smoothing, threshold dependence and localisation in cluster inference.
Neuroimage. 2009;44(1):83–98Ramsey J, Zhang J, Spirtes P.
Proceedings of the 22nd Annual Conference on Uncertainty in Artificial Intelligence (UAI-2006) Association for Uncertainty in Artificial Intelligence,
Arlington, VA; 2006. Adjacency-faithfulness and conservative causal inference; pp. 401–408
Return to B COMPLEX
Return to ALZHEIMER's
Since 5-26-2013
| Home Page | Visit Our Sponsors | Become a Sponsor |
Please read our DISCLAIMER |