FROM:
Proc Natl Acad Sci U S A. 2006 (May 2); 103 (18): 7130–7135 ~ FULL TEXT
Veronica Galvan, Olivia F. Gorostiza, Surita Banwait, Marina Ataie, Anna V. Logvinova, Sandhya Sitaraman, Elaine Carlson, Sarah A. Sagi, Nathalie Chevallier, Kunlin Jin, David A. Greenberg, and Dale E. Bredesen
Buck Institute for Age Research,
8001 Redwood Boulevard,
Novato, CA 94945, USA.
The deficits characteristic of Alzheimer's disease (AD) are believed to result, at least in part, from the neurotoxic effects of beta-amyloid peptides, a set of 39-43 amino acid fragments derived proteolytically from beta-amyloid precursor protein (APP). APP also is cleaved intracytoplasmically at Asp-664 to generate a second cytotoxic peptide, APP-C31, but whether this C-terminal processing of APP plays a role in the pathogenesis of AD is unknown. Therefore, we compared elements of the Alzheimer's phenotype in transgenic mice modeling AD with vs. without a functional Asp-664 caspase cleavage site.
Surprisingly, whereas beta-amyloid production and plaque formation were unaltered, synaptic loss, astrogliosis, dentate gyral atrophy, increased neuronal precursor proliferation, and behavioral abnormalities were completely prevented by a mutation at Asp-664. These results suggest that Asp-664 plays a critical role in the generation of Alzheimer-related pathophysiological and behavioral changes in human APP transgenic mice, possibly as a cleavage site or via protein-protein interactions.
Keywords: neurodegeneration, β-amyloid precursor protein-C31, β-amyloid precursor protein intracytoplasmic domain, caspase, memory
From the FULL TEXT Article:
Background
Alzheimer's disease (AD) is characterized by senile plaques, neurofibrillary tangles, and loss of synapses and neurons in the brain. The predominant proteinaceous component of senile plaques is β-amyloid (Aβ) peptide, and the “amyloid hypothesis” states that Aβ initiates the cascade of events that results in AD (1). Aβ precursor protein (APP) transgenic mice with high Aβ levels in the brain show synapse loss, behavioral changes, and synaptic transmission deficits before the formation of senile plaques (2, 3). APP also may be cleaved intracytoplasmically at Asp-664 by caspases (4, 5), liberating a cytotoxic carboxyl-terminal peptide, APP-C31 (5, 6).
One potential link between Aβ production and APP-C31 generation has been described, with the demonstration that APP mediates a significant component of Aβ cytotoxicity in cultured neural cells (7, 8). This finding suggests a model in which Aβ complexes with APP and induces APP multimerization, leading to cleavage of the APP cytosolic tail and initiating synaptic and neuronal damage (7). Thus, cleavage of APP at Asp-664 by caspases or caspase-like proteases may be a critical pathway mediating Aβ-induced cytotoxicity.
Despite these findings, the role (if any) that the intracytoplasmic cleavage of APP plays in vivo in AD pathogenesis is unknown. Therefore, we generated transgenic mice expressing an APP transgene used in an established mouse model of AD: platelet-derived growth factor B-chain promoter-driven APP transgenic mice (PDAPP) mice, which carry the familial AD-associated Swedish and Indiana mutations (2, 3), except that the C-terminal cleavage site in APP was mutated [Asp→Ala (D664A)]. The resultant mice were designated PDAPP(D664A) (aspartate to alanine mutation at position 664 of APP, D664A) mice.
Here we show that, although Aβ production and amyloid deposits are unaltered by Asp-664 mutation, Asp-664 is required for the pathophysiological and behavioral deficits characteristic of the AD phenotype. These results suggest that Asp-664 plays an important role in the generation of AD-like pathophysiology and behavior in human APP (hAPP) transgenic mice, possibly downstream of Aβ interaction with APP, either via cleavage at Asp-664 or via an intermolecular interaction (homomeric or heteromeric) requiring Asp-664.
METHODS
Generation of Transgenic Mice.
A G-to-C point mutation was introduced in the platelet-derived growth factor B-chain promoter-driven human APP minigene carrying the Swedish and Indiana mutations (2, 3) that mutated Asp-664 (APP695 numbering) to Ala [PDAPP(D664A)]. This mutation was confirmed by sequencing and by allele-specific DNA amplification (Applied Biosystems). Two rounds of transgenic injections were conducted. In the first, a 2 ng/µl solution of vector-free human PDAPP(D664A) transgene DNA was microinjected into B6D2F1/J eggs. In the second, the same DNA was microinjected into C57BL/6J eggs. Identification of founders was by PCR (primers shown in Supporting Text). Transgenic lines were maintained by crosses with C57BL/6J breeders (Charles River Laboratories).
Western Blotting.
Western blotting was done as described in ref. 6 by using α-APP CT15 antibodies (12). Details are included in Supporting Text.
Detection of Soluble Aβ.
Aβ levels in brain were assessed from CHAPS-solubilized lysates by immunoprecipitation, followed by Western blotting with 26D6 anti-APP antibody (Aβ1–12). Immunoprecipitates were fractionated on bicine-urea SDS/PAGE gels to resolve the Aβ 40 and 42 species (34). Aβ 40 and 42 were quantitated by ELISA (BioSource, Camarillo, CA).
Quantitation of Synaptophysin Immunoreactivity.
Fifty-micrometer vibratome brain sections were stained with α-synaptophysin antibodies (10 µg/ml; Chemicon, Temecula, CA), followed by FITC donkey anti-mouse IgG (1:400; Vector Laboratories), counterstained with propidium iodide, and imaged with a confocal microscope (Nikon PCM-2000) by using a ×100 objective and a ×2.7 digital zoom. Quantitation of synaptophysin-immunoreactive presynaptic terminals in CA1 stratum radiatum was performed by a modification of the stereological dissector method (2). Details are included as Supporting Text.
Volume Determinations.
Volume determinations were done by using imaris 3d (Bitplane) and manual Cavalieri analyses. Both methods are described in Supporting Text.
Quantitation of GFAP Immunoreactivity.
Fifty-micrometer vibratome sections were stained with α-GFAP antibodies (10 µg/ml; Chemicon) and Alexa594 donkey α-mouse IgG (1:1,000; Invitrogen), counterstained with DAPI, and imaged by using a ×20 objective (PCM-2000; Nikon). The total area of GFAP immunoreactivity in the medial portion of the DG was determined for each animal by using simple pci (Compix, Cranberry Township, PA).
Behavioral Testing.
The Morris water maze (20) was used to test spatial memory. All mice had normal motor and visual skills. Swimming ability was assessed with a straight water alley (15 × 200 cm) containing a submerged (1 cm) 12 × 12 cm platform. No differences were observed in swimming abilities between groups. The procedure described by Morris et al. (20) was followed. Working memory and motivation to explore novelty were tested by using the Y maze task and a novel object exploration task, respectively. Details are included in Supporting Text.
RESULTS
Generation of PDAPP(D664A) Mice.
 Table 1
Table 1
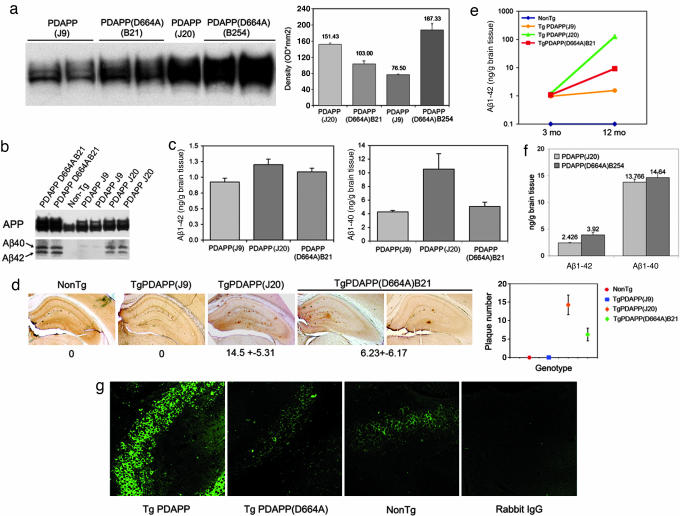 Figure 1
Figure 1
|
The D664A mutation was introduced into a hAPP minigene carrying the Swedish (K670N and M671L) and Indiana (V717F) familial AD mutations downstream from the platelet-derived growth factor B-chain promoter. The construct in which the D664A mutation was introduced was identical to that used in the generation of PDAPP mice, which represent a well established model of AD (2, 3). Transgenic animals generated from this construct were crossed onto the C57BL/6 background for 5 to 20 generations and compared with PDAPP transgenic mice (2, 3) in the same genetic background. Densitometric analyses of expression of the transgene showed that, among six PDAPP(D664A) lines generated, one line, designated B21, had APP expression levels between those of the low-expressor J9 and the high-expressor J20 line (Table 1 and Fig. 1a). Later, transgenic mice were generated from the same construct directly into the C57BL/6J background. Among 14 transgenic lines generated, a second PDAPP(D664A) line, designated B254, was selected, which demonstrated levels of expression of the hAPP transgene higher than those of the J20 line (Table 1 and Figure 1a).
To determine whether mutation of Asp-664 blocked C-terminal cleavage of human APP in vivo, we incubated brain sections from 3-month-old (mo) mice from all lines with an antibody that specifically recognizes the C-terminal neoepitope generated after cleavage of APP at Asp-664 (or Ala-664; APPNeo), and does not recognize full-length APP (4, 6). Although strong APPNeo immunoreactivity was detected in cell bodies and projections of hippocampal neurons in PDAPP mice, immunoreactivity in PDAPP(D664A) mice was indistinguishable from that observed in nontransgenic littermates of either transgenic line (Fig. 1g).
Lack of Effect of the D664A Mutation on Aβ Production and Deposition in Vivo.
Previous studies have shown that the D664A mutation does not affect Aβ production in cultured cells (12, 13), and similar results were obtained in vivo in the current study: We assayed for Aβ40 and Aβ42 in whole-brain lysates by immunoprecipitation and Western blotting (Fig. 1b) and quantitated their levels by ELISA (Fig. 1c). A summary of the comparison of PDAPP (J9 and J20) to PDAPP(D664A) (B21 and B254) mice is shown in Table 1. Levels of Aβ40 and 42 in young B21 mice measured by ELISA were intermediate between those of J9 and J20 lines, correlating with the levels of expression of the APP transgene in each transgenic line.
We next measured Aβ42 in 12-mo J9, J20, and B21 mice; again, J9 < B21 < J20 (Fig. 1e). Thus mutation of Asp-664 had no demonstrable effect on the net production of Aβ40 and 42 in vivo.
To determine whether Asp-664 has an effect on plaque deposition, we examined brain sections from 12-mo transgenic animals by using an antibody against Aβ (4G8). Quantitative determinations showed 4G8-immunoreactive Aβ deposits (J20 = 14.5 ± 5.3; B21 = 6.2 ± 6.1 plaques per section), restricted to the hippocampus and cortex. No amyloid deposits were found in J9 mice at 12 months (n = 7; Fig. 1d), but J9 mice did demonstrate plaques at 18–24 months (data not shown). Amyloid deposits in all three transgenic lines were thioflavin-S positive (data not shown). These results argue that mutation of Asp-664 in the intracytoplasmic domain of APP has neither a quantitative nor qualitative effect on the deposition of β-amyloid in vivo.
To exclude the possibility that the results obtained would be unique to the B21 transgenic line, we measured Aβ40 and 42 concentrations in J20 animals in comparison with a second, independent PDAPP(D664A) line, B254, whose levels of APP transgene expression are ≈20% greater than those of J20 (Fig. 1a). Quantitation of Aβ at 4 months in J20 and B254 brain lysates by ELISA showed that B254 had higher levels of both Aβ40 and 42 than J20, which were increased by 6% and 60%, respectively (Fig. 1f) (the reason for the disproportionate increase in Aβ42 in the B254 animals is not yet understood).
Effect of the D664A Mutation on Hippocampal Presynaptic Density Number.
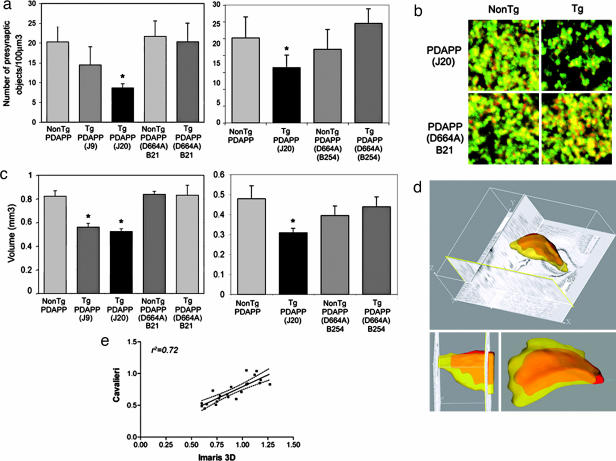 Figure 2
Figure 2
|
Decreases in the levels of synaptophysin in the hippocampus and prefrontal cortex correlate with cognitive decline in AD (14). PDAPP mice show decreased numbers of hippocampal synaptophysin-immunoreactive presynaptic densities (HSPDs) before plaque formation in a degenerative pattern (2, 3). To determine whether the APP cleavage (or interaction) at Asp-664 affects the loss of HSPDs in PDAPP mice, HSPDs were quantitated in the CA1 region of J9, J20, B21, and nontransgenic littermates at 8–10 mo by using the stereological dissector method (refs. 2 and 3; Methods). Both J9 and J20 mice displayed a reduction in the number of HSPDs in hippocampal CA1 (Figure 2a Left and Table 1). The reduction in HSPDs in J20 was ≈60%, and although we observed a consistent reduction (30%) in J9 animals, this difference did not reach statistical significance. Numbers of HSPDs in B21, however, were indistinguishable from controls (Fig. 2a). To exclude an insertional effect unique to B21, we measured HSPDs in B254 in comparison with J20 and controls. Although J20 showed a 45% decrease in HSPDs in this later study, B254 showed no decrease (Fig. 2a Right). Thus, the reduction in HSPDs observed in hippocampi of PDAPP mice was not present in PDAPP(D664A) mice.
Effect of the D664A Mutation on Astrogliosis.
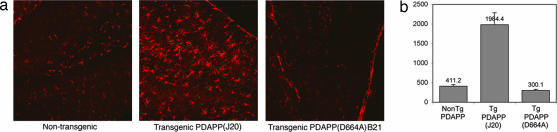 Figure 3
Figure 3
|
Astrogliosis occurs in many neurodegenerative diseases, including AD (15). Several mouse models of AD recapitulate this feature of AD (16). We stained hippocampal sections of brains from 12-mo transgenic mice with antibodies specific for glial fibrillary acidic protein (GFAP), a marker for astrocytes. A pronounced increase (4.5-fold) in GFAP immunoreactivity was observed in 12-mo J20 but not in B21 or control littermates (Figure 3). Thus, prevention of Asp-664 cleavage abolished astrogliosis in brains from hAPP transgenic mice.
Effect of the D664A Mutation on Dentate Gyrus Volume.
A decrease in cortical volume is one of the virtually constant neuropathological features of AD. Although not all models of AD have been surveyed for this feature, PDAPP mice display reduced dentate gyrus (DG) volumes at early ages (3–4 mo), especially in the molecular layer (17, 18). We therefore determined DG volumes in J9, J20, B21, and control littermates at 3-mo (18), both by digital 3D reconstruction of Nissl-stained sections and by manual Cavalieri analysis (ref. 17; Methods). DG atrophy was observed in brains of J9 and J20 but not B21 mice (Fig. 2 c and d). Volumes derived by the two methods were highly correlated (r2 = 0.72, P < 0.00001, n = 28; Fig. 2e). To exclude an effect unique to B21, we measured DG volumes in B254 in comparison with J20. Although J20 showed a 40% decrease in this later study, B254 showed no reduction (Fig. 2c Right). Thus, loss of DG volume in PDAPP animals is rescued in PDAPP(D664A) mice.
Effect of the D664A Mutation on AD-Associated Cognitive Abnormalities.
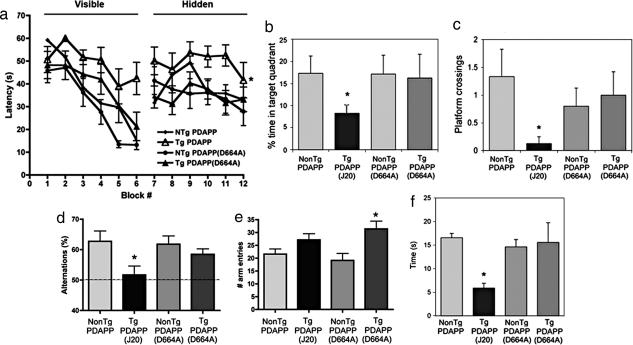 Figure 4
Figure 4
|
PDAPP mice demonstrate spatial learning and memory deficits (19) beginning at ≈6- to 7-mo. Therefore, we tested 12-mo J20, B21, and littermate controls in the Morris water maze (MWM) (20) after ensuring that motor and visual skills were intact. As previously described for J20 as well as other mouse models of AD (19, 21–24), MWM results showed that, although the majority of J20 learned to navigate to a visible and to a hidden platform (a significant effect of block number on performance, repeated-measures ANOVA; P < 0.0001; Figure 4a), their performance was impaired with respect to their nontransgenic littermates and to B21 in the hippocampal-dependent, spatial component of the task (two-way ANOVA; P < 0.0001). In the posttraining probe trial, J20 spent less time in the target quadrant (P < 0.05; Fig. 4b) and passed over the prior location of the platform significantly less often than controls (P < 0.02; Fig. 4c), whereas target crosses by B21 were not different from control groups. No differences in swimming speed were found among groups. To exclude an effect unique to B21, we tested spatial memory in B254 in comparison with J20. Although performance of transgenic J20 mice was impaired, B254 showed no significant deficits in the hidden task or in the probe trial (Supporting Text; see also Fig. 6, which is published as supporting information on the PNAS web site).
Some transgenic models of AD (23, 25, 26) show deficits in working memory at early ages (10–12 weeks). To evaluate working memory, we tested young (12-week-old) J20 and B254 animals in the Y maze. Alternations of entries in the arms of the Y maze were significantly reduced in J20, suggesting an impairment in working memory, but not in B254 (Fig. 4d). Spontaneous activity showed a trend toward increase in J20, and a significant increase in B254 transgenic animals (Fig. 4e). Thus, our results suggest that a defect in working memory in J20 animals is rescued by mutation of Asp-664 in the human familial AD-APP transgene. However, the D664A mutation did not rescue the increase in spontaneous activity associated with hAPP transgene expression.
Other behavioral abnormalities also have been described for mouse models of AD, such as neophobia (27), a decrement in the exploration time devoted to novel objects or regions. This behavioral pattern is associated with decreased glucose utilization in the entorhinal cortex, an age-related impairment exhibited by some PDAPP mice, aged nontransgenic mice, some cognitively impaired humans, and patients with AD (27). Because neophobia also is detectable in some mouse models of AD as early as 1 month of age (27), we compared J20 and B254 in a simple novel object exploration test at 3 months of age. J20 showed neophobia when compared with control littermates but B254 did not (Fig. 4f). These results suggest that the diminished time devoted to explore novel objects displayed by young PDAPP mice may require Asp-664 in the hAPP transgene.
Effect of the D664A Mutation on hAPP-Induced Enhancement of Hippocampal Neuronal Precursor Proliferation.
Recently, neurogenesis has been shown to be increased in the hippocampi of patients with AD (28) and in the brains of some, (29) but not all (30, 31), transgenic models of AD, and it has been suggested that this effect may be part of the response of neurons to chronic neural injury or neurodegeneration. Therefore, we assessed the effect of the D664A mutation on hAPP-induced hippocampal neuronal precursor proliferation in young (3-mo) and older (12-mo) PDAPP mice (combined J9 and J20), PDAPP(D664A)(B21), and control littermates. Mutation of Asp-664 abolished the increase in numbers of proliferating cells present in the subgranular zone of the dentate gyrus in both young and old transgenic PDAPP mice (Fig. 7, which is published as supporting information on the PNAS web site).
DISCUSSION
Consistent with previous reports, transgenic PDAPP mice exhibited DG atrophy, diminished hippocampal presynaptic densities, astrogliosis, enhanced neuronal progenitor proliferation, spatial and working memory deficits, and neophobia. Examination of these features of the AD-like phenotype in mice genetically matched to PDAPP mice except for the presence of a D→A mutation at position 664 in the intracytoplasmic domain of the hAPP minigene revealed a normalization of all these parameters. In addition, we (and our collaborators) have documented in refs. 10 and 11, in abstract form, an improvement in basal synaptic transmission associated with the D664A mutation. In contrast, soluble Aβ accumulation and amyloid deposition in PDAPP and PDAPP(D664A) mice were not significantly different. Our findings suggest that Asp-664 does not affect Aβ production but is critical for synaptic loss, DG atrophy, astrogliosis, synaptic transmission deficits, enhanced neurogenesis, and behavioral abnormalities in hAPP-transgenic mice. These features of the PDAPP mouse model of AD are likely to involve either APP cleavage at Asp-664 or protein–protein interactions (homomeric or heteromeric). Whether Asp-664 is critical for cleavage, protein–protein interactions, or both, these features may occur downstream from events initiated by Aβ or independently of Aβ effects (32) (the wealth of literature implicating Aβ in AD favors the former alternative conclusion over the latter).
Our data lend support to a recently proposed model of AD, in which Aβ binds to (7, 8, 33) and oligomerizes (7) APP, leading to cleavage at Asp-664 and cytotoxicity (7). Our results do not exclude the possibility that the Asp-664→Ala mutation affects the AD phenotype not by preventing cleavage at Asp-664 but rather by affecting an as-yet-uncharacterized protein–protein interaction; however, in either case, the mediation of Aβ toxicity in vivo by APP via an intracytoplasmic mechanism (be it cleavage or protein–protein interaction, or both) is supported by the current observations.
The C-terminal cleavage of APP by caspases truncates APP amino terminally to sequences required for its interaction with motor proteins, components of the stress response, and transcriptional transactivators. Cleavage of APP by transiently activated caspases at neuronal terminals, therefore, may disrupt its interaction with different protein complexes and, thus, alter the normal processing, turnover, or function of the molecule. Thus, it is possible that protective mechanisms that partially compensate for Aβ toxicity may be up-regulated when the C-terminal cleavage of APP is precluded.
The results presented here point to a key role for the C-terminal cleavage of APP (or alternatively, a protein–protein interaction requiring Asp-664) in the development of early structural and functional AD-like deficits in a transgenic mouse model. Furthermore, our data indicate that the intracytoplasmic domain of APP may play an important role in the pathogenesis of AD, suggesting that the cleavage of APP at Asp-664 may represent a therapeutic target.
ACKNOWLEDGMENTS
We thank Drs. Edward Koo, Brock Schroeder, and Alexei Kurakin for helpful discussions; Dr. Lennart Mucke (University of California, San Francisco) for the J9 and J20 transgenic mice; Dr. Junli Zhang for production of transgenic lines; Molly Susag for administrative assistance; and Adam Bredt, Aaron Bae, Diba Ataie, and Wei Huang for technical assistance. This work was supported in part by National Institutes of Health Grants NS45093 and AG05131, The Joseph Drown Foundation, a grant to the Buck Institute from American Bioscience, Inc., and NIRG-04-1054 from the Alzheimer's Association (to V.G.). V.G. thanks the John Douglas French Alzheimer's Foundation and Mrs. Eloise Goodhew Barnett for their support.
Conflict of interest statement:
No conflicts declared.
ABBREVIATIONS
Aβ = β-amyloid
AD = Alzheimer's disease
APP = β-amyloid precursor protein
D664A = aspartate to alanine mutation at position 664 of APP
GFAP = glial fibrillary acidic protein
hAPP = human APP
HSPD = hippocampal synaptophysin-immunoreactive presynaptic density
mo = month-old
PDAPP = platelet-derived growth factor B-chain promoter-driven APP transgenic
References
Selkoe D. J.
Science. 2002;298:789–791
Hsia A. Y., Masliah E., McConlogue L., Yu G.-Q., Tatsuno G., Hu K., Kholodenko D., Malenka R. C., Nicoll R. A., Mucke L.
Proc. Natl. Acad. Sci. USA. 1999;96:3228–3233
Mucke L., Masliah E., Yu G. Q., Mallory M., Rockenstein E. M., Tatsuno G., Hu K., Kholodenko D., Johnson-Wood K., McConlogue L. J.
Neurosci. 2000;20:4050–4058
Gervais F. G., Xu D., Robertson G. S., Vaillancourt J. P., Zhu Y., Huang J., LeBlanc A., Smith D., Rigby M., Shearman M. S., et al.
Cell. 1999;97:395–406
Lu D. C., Rabizadeh S., Chandra S., Shayya R. F., Ellerby L. M., Ye X., Salvesen G. S., Koo E. H., Bredesen D.
E. Nat. Med. 2000;6:397–404
Galvan V., Chen S., Lu D., Logvinova A., Goldsmith P., Koo E. H., Bredesen D. E. J.
Neurochem. 2002;82:283–294
Lu D. C., Shaked G. M., Masliah E., Bredesen D. E., Koo E. H. Ann.
Neurol. 2003;54:781–789
Lorenzo A., Yuan M., Zhang Z., Paganetti P. A., Sturchler-Pierrat C., Staufenbiel M., Mautino J., Vigo F. S., Sommer B., Yankner B. A. Nat.
Neurosci. 2000;3:460–464
Chin J., Palop J. J., Puolivali J., Massaro C., Bien-Ly N., Gerstein H., Scearce-Levie K., Masliah E., Mucke L.
J. Neurosci. 2005;25:9694–9703
Galvan V., Saganich M., Schroeder B., Gorostiza O. F., Logvinova A., Banwait S., Jin K., Greenberg D. A., Mucke L., Heinemann S., et al.
Society for Neuroscience. San Diego: ScholarOne; 2004
Schroeder B., Saganich M., Galvan V., Long J. M., Bredesen D. E., Heinemann S., Koo E. H.
Society for Neuroscience.
San Diego: ScholarOne; 2004
Soriano S., Lu D. C., Chandra S., Pietrzik C. U., Koo E. H. J.
Biol. Chem. 2001;276:29045–29050
Tesco G., Koh Y. H., Tanzi R. E.
J. Biol. Chem. 2003;278:46074–46080
Terry R. D., Masliah E., Salmon D. P., Butters N., DeTeresa R., Hill R., Hansen L. A., Katzman R.
Ann. Neurol. 1991;30:572–580
Streit W. J. J. Neurosci. Res. 2004;77:1–8
Irizarry M. C., Soriano F., McNamara M., Page K. J., Schenk D., Games D., Hyman B. T. J.
Neurosci. 1997;17:7053–7059
Dodart J. C., Mathis C., Saura J., Bales K. R., Paul S. M., Ungerer A.
Neurobiol. Dis. 2000;7:71–85
Redwine J. M., Kosofsky B., Jacobs R. E., Games D., Reilly J. F., Morrison J. H., Young W. G., Bloom F. E.
Proc. Natl. Acad. Sci. USA. 2003;100:1381–1386
Palop J. J., Jones B., Kekonius L., Chin J., Yu G.-Q., Raber J., Masliah E., Mucke L.
Proc. Natl. Acad. Sci. USA. 2003;100:9572–9577
Morris R. J. Neurosci. Methods. 1984;11:47–60
Westerman M. A., Cooper-Blacketer D., Mariash A., Kotilinek L., Kawarabayashi T., Younkin L. H., Carlson G. A., Younkin S. G., Ashe K. H.
J. Neurosci. 2002;22:1858–1867
Janus C., Pearson J., McLaurin J., Mathews P. M., Jiang Y., Schmidt S. D., Chishti M. A., Horne P., Heslin D., French J., et al.
Nature. 2000;408:979–982
Moran P. M., Higgins L. S., Cordell B., Moser P. C.
Proc. Natl. Acad. Sci. USA. 1995;92:5341–5345
King D. L., Arendash G. W., Crawford F., Sterk T., Menendez J., Mullan M. J.
Behav. Brain Res. 1999;103:145–162
Holcomb L., Gordon M. N., McGowan E., Yu X., Benkovic S., Jantzen P., Wright K., Saad I., Mueller R., Morgan D., et al.
Nat. Med. 1998;4:97–100
Holcomb L. A., Gordon M. N., Jantzen P., Hsiao K., Duff K., Morgan D.
Behav. Genet. 1999;29:177–185
Hsiao K. K., Borchelt D. R., Olson K., Johannsdottir R., Kitt C., Yunis W., Xu S., Eckman C., Younkin S., Price D., et al.
Neuron. 1995;15:1203–1218
Jin K., Peel A. L., Mao X. O., Xie L., Cottrell B. A., Henshall D. C., Greenberg D. A.
Proc. Natl. Acad. Sci. USA. 2004;101:343–347
Jin K., Galvan V., Xie L., Mao X. O., Gorostiza O. F., Bredesen D. E., Greenberg D. A.
Proc. Natl. Acad. Sci. USA. 2004;101:13363–13367
Haughey N. J., Nath A., Chan S. L., Borchard A. C., Rao M. S., Mattson M. P. J.
Neurochem. 2002;83:1509–1524
Sturchler-Pierrat C., Abramowski D., Duke M., Wiederhold K. H., Mistl C., Rothacher S., Ledermann B., Burki K., Frey P., Paganetti P. A., et al.
Proc. Natl. Acad. Sci. USA. 1997;94:13287–13292
Lu D. C., Soriano S., Bredesen D. E., Koo E. H. J.
Neurochem. 2003;87:733–741
Scheuermann S., Hambsch B., Hesse L., Stumm J., Schmidt C., Beher D., Bayer T. A., Beyreuther K., Multhaup G. J.
Biol. Chem. 2001;276:33923–33929
Weggen S., Eriksen J. L., Das P., Sagi S. A., Wang R., Pietrzik C. U., Findlay K. A., Smith T. E., Murphy M. P., Bulter T., et al.
Nature. 2001;414:212–216

Return to ALZHEIMER's
Since 4-14-2020
|



