

Impact of a Complex Nutraceutical Supplement on Primary
Tumour Formation and Metastasis in Trp53+/- Cancer-prone MiceThis section is compiled by Frank M. Painter, D.C.
Send all comments or additions to: Frankp@chiro.org


FROM: Mutagenesis. 2014 (May); 29 (3): 177–187 ~ FULL TEXT
Vadim Aksenov, Douglas Boreham1 and C. David Rollo
Department of Biology and Department of Medical Physics and Applied Radiation Sciences,
McMaster University,
1280 Main St. W., Hamilton,
Ontario L8S 4K1, Canada
A complex dietary supplement designed to impact multiple mechanisms associated with aging and cancer reduced overall tumorigenesis in cancer-prone heterozygous Trp53+/- mice by ~30% (P < 0.018). Carcinomas were reduced by 67% (P < 0.006). Remarkably, metastasis (a leading cause of cancer mortality) was undetectable in treated animals (P < 0.004), and the occurrence of multiple primary tumours was reduced by 74% (P < 0.012). Reduction of pulmonary adenocarcinoma by 62% (P < 0.021) was of particular note given that lung cancer is the second leading cause of death in humans. Tumours showed pronounced age-related expression in untreated animals older than 600 days. Benefits of treatment only emerged in these later ages, suggesting that the supplement acted on mechanisms common to aging and cancer. The supplement was administered daily on bagel bits that were usually eaten within minutes by the mice. Although longevity was not statistically different between treatments, longevity was strongly related to the compliance of mice in eating the supplement. Linear regression revealed a strong positive relationship between the proportion of supplement eaten and the longevity of mice within the treatment group (P < 0.0001).
From the FULL TEXT Article:
Introduction
The p53 pathway crucially protects vertebrates from cancer.[1–3] Some 50–80% of human tumours are linked to loss of p53 function. [1-6] The p53 protein permits cell cycle progression under favourable conditions, induces cell cycle arrest if repair is required or initiates apoptosis if damage is severe. [3, 6] Signalling by p53 responds to diverse stressors including nutrient deprivation, hypoxia, oncogene activation and DNA damage. [3, 6] Disruption of p53 allows damaged cells to proliferate and progress to transformation. [1, 3] An important role for p53 in cell senescence highlights a trade-off between tumour suppression and aging. [1, 3, 6–8]
Mice with defective p53 are useful models for cancer studies. Tumour suppression depends on p53 expression, and consequently, mice with total p53 disruption have more severe tumorigenesis than those haploid for one functional p53 gene. [2, 3, 5, 7] The lifespan of knockout (Trp53–/–) mice is drastically reduced as they succumb to cancers within 10 months (2). Alternatively, Trp53+/– heterozygous mice exhibit delayed tumorigenesis manifesting at 1–2 years (and some escape cancer). [2] Thus, maximal longevity of Trp53+/– mice can be comparable to controls (~3 years) although mean survivorship is typically reduced as ~90% of Trp53+/– mice die by 2 years of age. [2] Heterozygous Trp53+/– mice are a better model for testing cancer interventions than Trp53–/– mice since severe tumorigenesis may overwhelm potential treatments and limit expression of cancers to only the earliest lethal forms. Delayed cancer in Trp53+/– mice may better resolve longer term treatment effects on a greater range of cancer types.
Cancer is the second leading cause of death in industrialised nations. [9, 10] Over 1.6 million new cases and over half a million cancer deaths were projected in the USA for 2012 alone. [10] The probability of developing cancer strongly increases with age with ~80% of cancer-related mortality occurring in those over 60 years old. [10] We found that cancer was also much greater in older cohorts of Trp53+/– mice indicating agerelated trends comparable to humans. Further, there is substantial overlap in the types of cancers in humans and Trp53+/– mice. The four cancers commonly affecting Trp53+/– mice are lymphomas, soft-tissue sarcomas, osteosarcomas and lung carcinomas. [2, 7] Lymphomas and lung carcinomas are among the 10 most frequent human cancers, and soft-tissue sarcomas are relatively common (10). One exception is osteosarcoma, which is frequent in Trp53+/– mice but rare in aging humans. Given the pervasive dysregulation of p53 in human and murine cancers, the similarity of cancer types and comparable age-related trends, Trp53+/– mice provide reasonable models for general understanding of cancer. Although humans are intrinsically more resistant to cancer than inbred mice, benefits ameliorating fundamental mechanisms like p53+/– may translate to humans.
Consideration of dietary supplements (DSPs) for preventing or treating cancer grows exponentially. Searching Google Scholar for ‘dietary supplements + cancer + prevention’ returns 3,500 articles for 2012 alone. The possible effectiveness of nutraceuticals (particularly antioxidants) is highly controversial. [11, 12] Many studies find little benefit although a recent clinical trial found definitive evidence for amelioration of cancer by a commercial multiple vitamin. [13] Another study found that participants ingesting high amounts of vitamins E and C, selenium and zinc were 67% less likely to develop pancreatic cancer. [14] Inconsistent results may reflect variable experimental designs (particularly the length of treatment and follow-up periods), diverse confounding factors, potential prooxidant properties of antioxidants and lumping DSPs into a single category despite enormous functional diversity. Against this background, meta-analyses may paint effective treatments with the stigma of majority failure. Furthermore, in vitro findings often fail to translate to whole organisms.
Here, we show that a complex DSP designed to target multiple mechanisms associated with aging and cancer significantly reduced primary and metastatic tumorigenesis in Trp53+/– mice. Effectiveness likely reflects a design targeted to multiple mechanisms and synergistic, complementary and emergent interactions among ingredients.

Table 1 The DSP employed here was based on our original formulation designed to target five key mechanisms of aging. This modestly extended longevity and strongly ameliorated age-related declines in cognition, sensory systems and motor functions in mice. [15–19] Given the strong age-related aetiology of many cancers [3, 6, 10], we hypothesised that a well-designed combination of ingredients might also impact cancer development. Consequently, we expanded our original formulation [17] to include five new ingredients relevant to tumorigenesis (Table I). Here, we report significant impacts of this DSP on diverse tumour types and explore likely mechanisms.
Materials and methods
Animals
A total of 114 heterozygous p53 (Trp53+/–) male mice were studied (F1 progeny of Jackson Trp53+/– males x SVJ129 +/+ females). Genotype was confirmed by Mouse Genotype LLC, Carlsbad, CA. All protocols adhered to McMaster University and the Canadian Council for Animal Care standards. Mice were housed three per cage in specific pathogen-free conditions. Aggression between cage-mates was rare, and aggressive mice were separated. Standard rodent chow (Teklad® 8640) and water were provided ad libitum. Facilities were maintained at 23°C ± 2°C with a 12:12 h light:dark cycle.
Complex DSP and treatment protocol
Table I lists components and dosages of the complex DSP. A review of the properties of all 35 ingredients relevant to cancer is precluded here. The original ingredients were targeted to offset oxidative stress, insulin resistance and inflammation while promoting mitochondrial function and membrane integrity. [17–19] These are all variously relevant to cancer. [20] We added five ingredients highlighted for their anti-cancer impacts (curcumin, resveratrol, lycopene, quercetin and pomegranate). [21] Dosages were calculated from high-end recommendations for humans adjusted for the smaller size and higher metabolism of mice. [15]
Curcumin activates p53 and can inhibit proliferation, metastasis and angiogenesis of cancers. [22–24] It has antioxidant and anti-inflammatory activity and inhibits the cell cycle, metalloproteinases and multiple growth factors via inhibition of signal transducer and activator of transcription (STAT) signalling. [22, 23, 25] Curcumin also inhibits activator protein 1 and promotes apoptosis via inhibition of nuclear factor-κB (NF-κB). [23, 26] Resveratrol impacts are similar to curcumin (antioxidant protection, p53 and apoptotic activation, inhibition of STAT growth pathways and NF-κB). It also enhances insulin sensitivity. [24,27–30]Lycopene is an antioxidant that inhibits growth factors, NF-κB and promotes apoptosis. [21, 31–33] Quercetin has antioxidant properties and inhibits the cell cycle and metalloproteinases. It inhibits growth factors including epidermal growth factor crucial to many cancers and promotes apoptosis via activation of FOXO1. [34–37] Pomegranate can induce apoptosis in cancer cells via inhibition of insulin-like growth factor-1 (IGF-1) signalling (and other growth factors) highlighted in cancers. It also has antioxidant and anti-inflammatory properties and inhibits NF-κB. [38–40]
Of the 114 mice, 60 were randomly assigned to the DSP-supplemented group and 54 remained untreated. In previous studies, we began treatment at weaning [15–19], but here, mice were obtained at ages between 75 and 115 days (mean age = 98 days) and supplementation was initiated immediately. The DSP (see below) was prepared by mixing all ingredients with distilled water and pipetting the combined slurry onto a small bread (bagel) bit (1 × 1 × 0.5 cm). Supplemented mice received one dose each day in their home cages. Untreated mice received just a bagel bit of standard size.
Some mice were initially reluctant to eat the entire bagel square and full dose. Therefore, we began by feeding mice a plain bagel square (which they consumed avidly) followed by gradually increasing the amount of DSP soaked onto the bagel square over a 3-month period. As a result, supplementation with a full dose was not achieved until ages of 165–205 days (~6 months). All mice were then maintained until endpoint.
Dietary consumption index
Despite gradual acclimation, some mice did not consistently eat the entire dose. Non-compliant mice were housed separately, so their consumption could be monitored. We also periodically isolated other mice to obtain a consumption index for all mice. We checked for uneaten DSP remnants immediately following each feeding session (most mice avidly consume the entire bagel bit and DSP within a few minutes). The weight of any uneaten piece was subtracted from that of the initial full square. We compiled a consumption index for each mouse as a score between 1 and 0 (where 1 indicates 100% consumption, 0.75 indicates 75% consumption, etc.). We applied regression analysis to determine the relationship between compliance and longevity.
Longevity
Longevity of control and treated Trp53+/– mice was assessed by analysing lifetime survivorship curves. Animals found dead or culled due to tumour endpoints were included in the survivorship analysis. In eight young treated animals and two untreated mice, endpoint was a severe ulcerative dermatitis. This reflects sporadic autoimmunity characteristic of C57BL/6 mice that is unrelated to cancer. [41] These were excluded from the survivorship analysis.
Body mass
Mice were periodically weighed between 75 and 500 days of age. Each mouse was weighed on 10 occasions unless it died prior to 500 days. Growth rate was assessed via regression analysis of mass against age for mice <300 days old. Body mass plateaued at ages >400 days, and we compared this mature body size between treatments with a t-test.
Pathology
Mice were sacrificed at endpoints that included development of a large tumour, limb paralysis, poor body condition, immobility, frailty, irregular breathing and prolapses. Of the 114 mice, 81 were suitable for complete necropsy and histological assessment. Five mice that were not necropsied but that expressed large abdominal tumours (characteristic of soft-tissue sarcoma) were included in the ‘total cancer’ analysis.
Standard tissues (sternum, thymus, heart, lungs, liver, spleen, kidneys, adrenals, thoracic spine, lumbar spine and brain) and any abnormalities (tumours, enlarged organs, lymph nodes, etc.) were collected. All tissues were fixed in 10% buffered formalin. Vertebrae and other mineralised tissues were further processed in an EDTA (145 g/l) solution.
Trimmed fixed tissue sections were embedded in paraffin. Paraffin blocks were sectioned on a Leica RM 2165 microtome at 3-µm thickness and stained with hematoxylin and eosin for histological examination. The presence of cancer, tumour types or other disease was diagnosed by an expert veterinary pathologist (Dr Dean Percy, Guelph, ON, Canada) based on slide examination and consultation of necropsy reports. Blind repeat samples were resubmitted for quality assurance with a 100% demonstrated precision record. Full pathology reports were obtained for 38 of 54 untreated mice and 43 of 60 supplemented mice. Since the proportion of animals that we failed to collect tissues from was virtually identical between treatments and causes were random, the untested animals did not introduce any experimental bias.
Statistical analyses
Total cancers in supplemented and untreated mice and each specific type of tumour were compared using a two-tailed chi-square (Fisher’s) test. Analysis of tumour types was based strictly on mice that had complete necropsy examination. The percent reduction of tumorigenesis for each cancer type was calculated as follows:Percent reduction = 100 – (Treated/Untreated),
where ‘treated’ is percent occurrence in treated animals and ‘untreated’ is percent occurrence in untreated mice.
Differences in survivorship between treatments were analysed with the logrank test. Growth rate was compared with regression (homogeneity of slopes) analysis. Mature body mass was compared using a t-test. Regression analysis was used to test the correlation between longevity and the DSP consumption index. All analyses were performed using Statistica® 6.0 software.
Results
Spontaneous tumorigenesis
Table 2 The number of mice succumbing to tumours was significantly reduced by the DSP. Of 41 control mice, 34 (83%) expressed tumours compared with only 26 of 45 supplemented mice (58%) (30% reduction, P < 0.018, Table II). The predominant tumours were sarcomas, lymphomas and carcinomas. Tumour incidences with respect to treatments are shown in Table II.
Sarcoma
Occurrence of sarcomas was most frequent, affecting 44% of animals. Osteosarcoma was found in 13 untreated mice (34%) and 8 supplemented mice (19%). The 44% reduction in incidence of osteosarcomas in supplemented mice fell short of significance (P < 0.132, Table II). Osteosarcomas were predominantly observed in lumbar vertebrae, but occasionally, the skull, jaw, liver, lung and sternum were affected. Soft-tissue sarcomas were observed in 16% of mice (22% if the five mice that were not necropsied are included). Of necropsied mice, hemangiosarcoma affected three untreated and five supplemented mice. Two cases of fibrosarcoma were diagnosed in supplemented mice and one case in an untreated mouse. One case of histocytic sarcoma affecting seven tissues was found in an untreated mouse. One case of astrosarcoma (brain) and one case of chondrosarcoma occurred in supplemented mice. No significant differences were obtained in any of these comparisons.
Carcinoma
Carcinoma affected 27% of animals and was diagnosed in 16 untreated and 6 supplemented mice (Table II). DSP treatment resulted in a 67% decrease in carcinoma (P = 0.006, Table II). Other than two cases of basosquamous cell carcinoma in untreated mice, the rest were pulmonary adenocarcinomas. Frequency of pulmonary adenocarcinomas was 37% in untreated mice (14 of 38) compared with only 14% in supplemented mice (6 of 43). The 62% reduction was statistically significant (P = 0.021; Table II).
Lymphoma
Lymphomas occurred in 17% of animals, affecting five supplemented and nine untreated mice (Table II). Due to sample size, this 50% reduction was not statistically significant. In all but one case, lymphomas were extra-nodal affecting a wide range of tissues, including lung, liver, spleen, kidneys and vertebrae. Often, multiple tissues and organs were affected in the same animal.
Pheochromocytoma
Two cases of pheochromocytoma were diagnosed in untreated mice. Although this tumour was not found in supplemented animals, low incidence precluded statistical significance.
Multiple tumour burden
Multiple tumour types of primary origin were found in 13 untreated mice (34%), most frequently a combination of pulmonary adenocarcinoma and osteosarcoma. In two cases, three primary tumour types were present. Conversely, only four supplemented mice (9%) had multiple tumours (Table II). A chi-square test confirmed that the incidence of multiple primary tumours was significantly lower in supplemented animals (P < 0.012; Table II).
Metastasis
Metastases were confirmed in seven untreated mice (18%). Remarkably, tumours of metastatic origin were absent in supplemented animals. This difference was highly significant (P < 0.004, Table II), indicating that the DSP effectively limited metastatic cancer.
Relationship between tumorigenesis and age
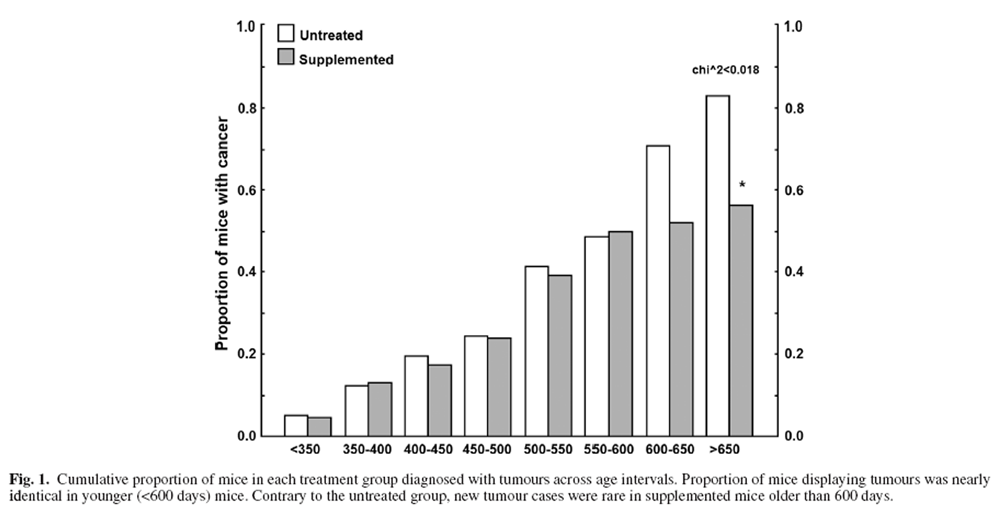
Figure 1 Mice sacrificed due to tumour-related endpoints were categorised into 50-day intervals, and age-related tumour burden is presented in Figure 1. The youngest mouse to develop a tumour was 249 days old. Between 350 and 600 days of age, roughly 2–6 mice were diagnosed with a tumour every 50 days. Tumorigenesis was similar for supplemented and untreated mice prior to 600 days of age (Figure 1) but rose steeply in untreated mice >600 days old compared with little increase in supplemented mice (Figure 1). Thus, benefits of the DSP mainly manifested in older ages.
Survivorship and dietary supplementation
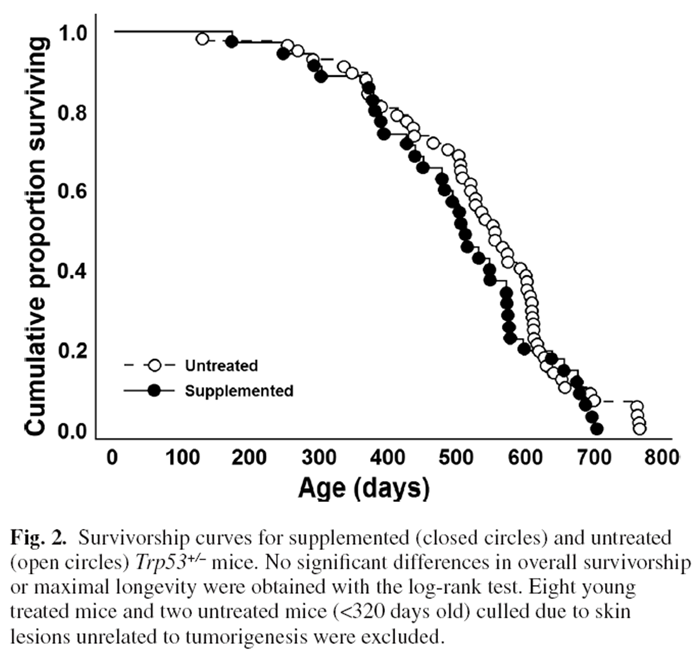
Figure 2
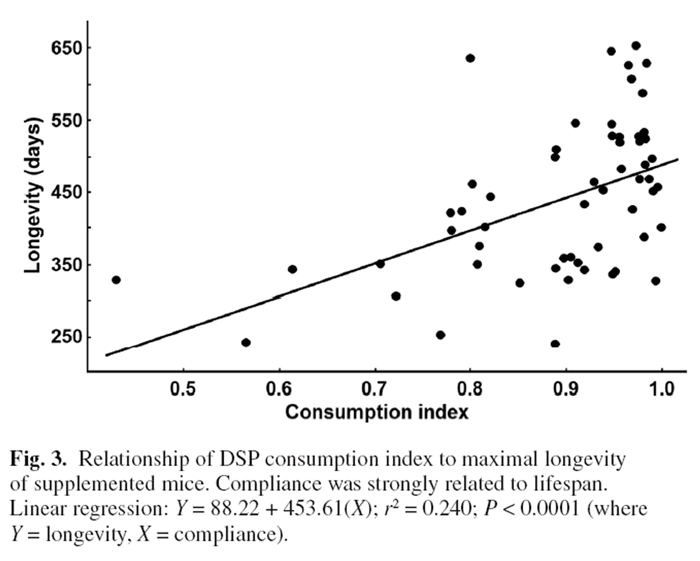
Figure 3 Surprisingly, log-likelihood analysis did not detect a significant difference in average survivorship between DSP and control mice (Figure 2) despite the marked reduction in lifetime tumour frequency. This likely reflected that there was little difference in tumorigenesis between treatments prior to 600 days with large impacts of the DSP emerging only in the oldest age cohorts. Several untreated mice that fortuitously did not develop cancer lived the longest (but maximal longevity did not differ significantly for a t-test of the last 10% of surviving mice). Eight younger supplemented and two untreated mice were terminated as they developed ulcerative skin lesions. Such lesions are common and specific to the C57BL/6 mouse strain.
They reflect sporadic autoimmune disease unrelated to cancer. [41] Regardless, longevity was strongly linked to rates of DSP consumption. Two mice had a DSP consumption index of <0.6; 85% of mice had an index of 0.8 or better; and 62% and 41% had indices of >0.9 or >0.95, respectively. Plotting the longevity for each supplemented mouse against its consumption index revealed a strong positive relationship between longevity and consumption of the DSP (P < 0.0001, Figure 3). The longest lived mice were those consistently ingesting the highest doses, whereas the shortest lived mice ate the least DSP (Figure 3).
Body mass
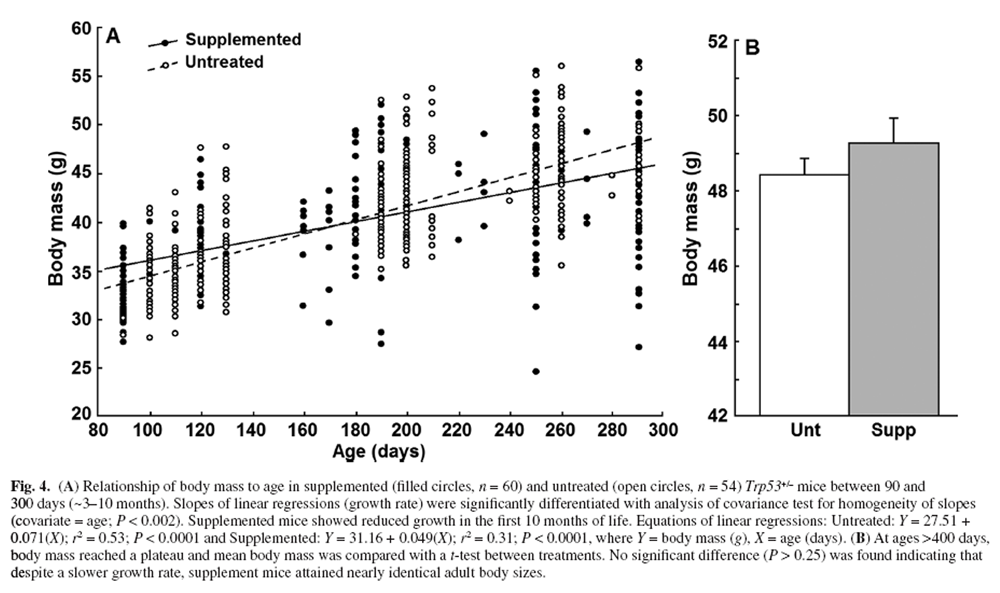
Figure 4 We regularly recorded body mass from 90 to 500 days of age. Figure 4A illustrates that the growth rate of mice <300 days of age was significantly higher in untreated compared with supplemented mice (linear regression, homogeneity of slopes analysis, P < 0.002). Comparison of mean adult body mass between treatments (mice >400 days old) with a t-test was not statistically significant (P > 0.25, Figure 4B). Despite significantly slower growth in DSP mice, body mass between the treatment groups at 290 days differed by only 3.6%.
Discussion
Overview of tumorigenesis
The DSP reduced the number of Trp53+/– mice succumbing to tumours by ~30% (P < 0.018, n = 86; Table II). The biggest reduction (67%) was in carcinoma (P < 0.006). Despite clear trends for other cancers, low sample sizes obscured statistical significance. For example, lymphomas affected only 17% of mice, so despite a 50% reduction in treated mice, impacts of the DSP proved not significant (Table II). Pheochromocytomas occurred exclusively in untreated mice but only two cases were diagnosed (Table II). Osteosarcomas were found in 26% of mice, but despite being 44% less frequent in supplemented mice, this also fell short of significance (P = 0.132; Table II).
A third of untreated Trp53+/– mice expressed multiple primary tumours (usually adenocarcinoma and osteosarcoma) (Table II). Conversely, multiple primary tumours afflicted only 9% of supplemented mice (a reduction of 74%, P < 0.013; Table II), strongly supporting effectiveness of the DSP against tumorigenesis. This may be particularly relevant to therapeutic applications where treatment of multiple tumours is challenging.
Cancer types in Trp53+/– versus normal mice
Sarcomas, lymphomas and carcinomas collectively comprise >95% of tumours occurring in mice. [2, 7] However, Trp53+/– mice exhibit significantly different proportions of tumours compared with normal mice. Thus, sarcomas typically comprise 20% of tumours in normal controls but can exceed 60% in Trp53+/– mice. [2, 7] We did not examine normal mice but we did find a high frequency of sarcomas (50%) in untreated Trp53+/– mice (Table II). In general, lymphoma and carcinomas are only 2- to 4-fold higher in Trp53+/– mice compared with normal controls (i.e. loss of p53 has a relatively small impact on frequency of these tumours), whereas sarcomas can be elevated 15- to 30-fold. [7]
Despite a hopeful trend for osteosarcoma, sarcomas generally were unresponsive to supplementation (20% reduction by the DSP, P = 0.378). Our overall estimate of effectiveness of the DSP (~30%) included sarcomas. Excluding sarcomas, the frequency of all other cancers in mice amounted to 71% of untreated mice, whereas DSP treatment reduced this to 26% (a 40% reduction, P < 0.0002). Considering that normal animals exhibit far fewer sarcomas and more carcinomas and lymphomas (that responded better to the DSP), DSP benefits would likely be relatively greater in normal mice with intact p53. In humans, sarcomas are also less frequent than lymphomas and carcinomas are similarly resistant to treatment. [10]
Lung cancer
Lung cancer comprised 14% of cancers diagnosed in the USA in 2012 but accounted for ~29% of cancer deaths, as lung cancer is refractive to treatment. [10] Pulmonary adenocarcinoma is the most frequent lung cancer worldwide and rates are increasing. [42] Remarkably, the DSP obtained 62% reduction in pulmonary adenocarcinoma in mice (P < 0.021, Table II). This is one of our most promising results given the high morbidity of this disease. Even modest translation to humans would yield large economic and social benefits.
Metastasis
Suppression of metastasis by the DSP (Table II, P < 0.004) is particularly important given that radiation or chemotherapy is required for treatment [43, 44] and malignancy contributes to 90% of cancer deaths. [45] Several aspects of cell transformation and angiogenesis are regulated by redox, suggesting an antioxidant mechanism for our results. [46] Likely contributors include matrix metalloproteinases (MMPs) and growth factors, such as IGF-1 [47–49], transforming growth factor-β and vascular endothelial growth factor (VEGF). [46,50,51] All are favoured by oxidative conditions. [46, 50, 52, 53] Inflammatory cytokines also contribute to MMP expression [54] and stimulate NF-κB, STAT [55] and VEGF. [56] Nearly one-third of cancer patients exhibit metastasis at first diagnosis. [57]
Cancers obtain high evolvability via an elevated oxidative milieu and strong survival signal. This facilitates adaptation to the host and treatments via complex alterations in cellular regulatory networks. Strong protection against radiation-induced DNA damage [58] by our original DSP suggests that mutagenesis essential for cancer progression and metastasis may be significantly reduced. Indeed accelerated growth of tumour cells with p53 deficiency was suggested to reflect increased mutagenesis and malignant progression. [59] Inhibition of metastasis by the DSP could extend the period for effective surgical intervention. [45] Interestingly, aspirin (in the DSP) reduces metastasis in humans. [60]
Aging and cancer
Cancers in untreated Trp53+/– mice showed strong age-related expression, whereas the increase in mice older than 600 days was much less in treated animals (Figure 1). The original DSP was designed to modulate five key mechanisms associated with aging (reduce free radical processes, ameliorate inflammation, increase insulin sensitivity, improve mitochondrial function and maintain membrane fluidity). [15–18] Age is the primary risk factor for many cancers [2, 10], and success of the DSP likely involves mechanisms common to both aging and cancer. [1, 3, 6] Full-dose supplementation was only achieved by ~6 months of age when some tumorigenesis could have been initiated. Better results may have been obtained with earlier intervention. Tumours take time to attain detectable size [61], and some mice reached endpoint at 250–350 days of age. Regardless, treatment benefits were most apparent in older ages that represent the most relevant cohort for assessing impacts. Mutation accumulation with age is undoubtedly important, but aging of the organismal/cellular environment [e.g. signalling, redox, senescent cells, functional declines (proteasome, autophagy, DNA repair)] could also impact cancer. [62]
Oxidative stress, the genome, cellular damage and tumorigenesis
The free radical theory of aging faces challenge, but there is a clear causal role of oxidative stress in cancer. Most spontaneous tumours trace to DNA damage and mutations driven by genotoxic stress (particularly reactive oxygen and nitrogen species [ROS]). [1, 3, 6, 45, 63–66] Several antioxidants and DNA repair mechanisms are regulated by p53. [59, 67] If DNA damage is detected, p53 arrests the cell cycle and promotes repair. If damage is severe, p53 promotes apoptosis or transition of cells to a senescent phenotype. [1, 3, 6] Reduction of DNA damage is likely to ameliorate cancer [31, 64, 68, 69], whereas low manganese superoxide dismutase (MnSOD) activity promotes cancer. [70]
Although the status of specific antioxidant enzymes and redox strongly vary among cancers and their stage of development, imbalanced antioxidant function and an oxidative milieu characterise most cancers. [71, 72] Overexpression of mitochondrial catalase ameliorated breast cancer in mice [73], including metastasis as in our study. N-acetylcysteine also strongly ameliorated cancer in Trp53–/– knockout mice. [59] Of the 35 ingredients in our DSP (including N-acetylcysteine), 27 scavenge diverse mitochondrial and cytoplasmic ROS, including H2O2, O2–, OH–, NO, NO–, O– and ONOO– . [17–19,58]
Chromosomal aberrations and oxidative DNA base damage are associated with cancer [63, 64, 74], and our original DSP prevented damage by 2 Gy of whole body ionising radiation (likely by rapid scavenging of radiation-induced free radicals). The DSP also reduced radiation-mediated apoptosis of lymphocytes, likely by preventing damage. [75] We further showed that oxidative and nitrosative protein damage in brains of supplemented mice were significantly reduced [17, 19], whereas oxidised and nitrated proteins are elevated in cancers. [65] A multiple DSP (23 vitamins, minerals and antioxidants) reduced oxidative DNA damage in lymphocytes of humans 45–70 years old [76], suggesting promise for our DSP.
Some debate regarding the free radical theory of aging reflects that some radicals are essential signalling molecules, whereas others, such as peroxynitrite, are highly damaging. Our original DSP ameliorates nitrosative stress even though this was not a design target. [19] Strong antioxidant activity of the DSP could ameliorate DNA damage, mutations and tumorigenesis across the lifetime of mice. [66] Aging and cancer may also reflect oxidative impacts on aspects such as mitochondrial integrity and biogenesis, proteasome function and autophagy. The DSP could protect these and redox-regulated ion channels supporting cellular functions. [77, 78]
Normally, superoxide generated by mitochondria is removed by MnSOD. [79] However, age-related accumulation of damage can impact electron transport chain (ETC) complexes [17, 18, 80, 81], leading to reduced energy and increased ROS generation. [79] Thus, maintaining mitochondrial function could reduce oxidative stress, DNA damage and mutagenesis. Several DSP ingredients support mitochondrial function [58], and our original DSP increased activity of mitochondrial ETC complexes II–IV and reduced oxidative damage. [17–19]
Long-lived calorically restricted (CR) mice express low levels of ROS [82, 83] via decreased mitochondrial ROS generation. [82–84] This may contribute to reduced tumorigenesis. [85] The DSP mimics some aspects of CR with respect to mitochondria [17–19] implicating improved mitochondrial function in the amelioration of cancer achieved here.
Inflammation and cancer
Inflammation was a design criterion for the original DSP [58] and is implicated in both aging and cancer. [86–88] Inflammation can impair antioxidant defenses [86] and increase ROS stress by activating NAD(P)H oxidases, nitric oxide synthase, myeloperoxidase, eosinophil peroxidase [88] and 5-lipoxygenase. [89] Nicotinamide adenine dinucleotide phosphate oxidase (in macrophages and neutrophils) and 5-lipoxygenase (in lymphocytes) are activated by inflammatory cytokines such as interleukin-1β [90] and can generate large amounts of ROS. [89] This can in turn induce further inflammatory cytokines. [50] Inflammation-mediated ROS can exacerbate mutagenesis and cancer. [88] Interleukin-6 is particularly important in cancer as it can promote proliferation and anti-apoptotic protection to cells expressing its receptor. [12] Anti-inflammatory drugs protect against cancer. [91–94]Hence, anti-inflammatory agents (including aspirin and antioxidants) in the DSP may contribute to reduced tumorigenesis in Trp53+/– mice (Table II).
Insulin resistance and tumorigenesis
Insulin resistance commonly develops with age [95, 96] and contributes to age-related pathologies including cancer. [97–99] There are 11 ingredients in our DSP that promote insulin sensitivity [58], and the original DSP lowers blood glucose and improves glucose clearance (S. Matravadia and V. Aksenov, unpublished theses). The mechanisms linking insulin resistance to cancer are unclear, but the anti-cancer impacts of aspirin, the diabetes drug metformin and resveratrol likely involve upregulation of the key energy sensor, AMP-activated protein kinase (AMPK). [100, 101] Signals of low energy (activated AMPK) can inhibit target of rapamycin (TOR) and upregulate FOXO. [102, 103] Although such actions can improve mitochondrial function [101], our DSP appears to increase energy supply. The DSP may act via diverse mechanisms such as increasing spontaneous exercise [17] and elevated stress-resistance and repair mechanisms (e.g. proteasome activity, antioxidant recharging and DNA repair).
Longevity, cancer and supplementation
Survivorship curves did not differ significantly between supplemented and control mice (Figure 2). Given that DSP impacts were largely restricted to mice >600 days old and the log-rank test is a measure of general health across the lifespan, this may not be surprising. Also, the lack of significant extension of maximal lifespan does not reflect negative consequences of the DSP since mice showed a strong positive relationship of longevity with compliance, and untreated Trp53+/– mice that escaped cancer can achieve normal life spans (Figure 2). Aging and cell senescence are regulated by p53 [3, 6, 7, 104] which may could account for lack of longevity extension by the DSP in this strain, whereas a small increase in longevity was previously obtained in normal outcrossed mice (Figure 2). [16]
DSP compliance and survivorship
Some mice did not consume a full dose of DSP on a daily basis. Forty-one percent of mice consistently ingested >95% of their daily dose and >85% of mice ingested at least 80% of the full dose over their lifetime. Previous studies with our original DSP obtained full compliance. [15–19] Greater bulk and taste of the cancer DSP likely impacted compliance. Most early deaths of supplemented mice were those showing poor compliance for ingesting the supplement. Thus, of six mice with a consumption index <0.78, none lived longer than 400 days compared with >700 days for some mice with compliance ratios close to 1.00 (Figure 3). As suggested by a reviewer, we re-examined the relationship between longevity and consumption index with the six poor eaters removed (<0.78 index). The regression for the remaining mice remained highly significant (P < 0.005) confirming that the relationship reported in Figure 3 was not artificially inflated by outlying points. Given that cancer is the main determinant of longevity for this strain, the significant benefit of the DSP on survivorship of Trp53+/– mice reinforces the anti-cancer actions of the diet.
Body size, supplementation and p53
The p53 pathway negatively impacts growth and proliferation via engaging cell cycle arrest. [1] Mice with amplified p53 expression have reduced body weight and diminished tumorigenesis (i.e. body mass is negatively associated with p53 expression). [7, 104] Remarkably, the size range for male C57BL/6 mice (Jackson Laboratory Phenotype Information) is generally <35 g, but our Trp53+/– mice attained mature sizes of >48 g (Figure 4). The DSP had a significant but small impact on growth rates, but no change in adult mass was obtained (Figure 4). This suggests that the DSP did not act via a dietary restriction mechanism. Curcumin and resveratrol (both in our DSP) can increase p53 expression and nuclear translocation [24] suggesting that the reduction in early growth rate could reflect p53 impacts.
Impacts on growth, however, were small and could just as well reflect reduced oxidative conditions. Activity of growth-promoting pathways (e.g. growth harmone and TOR) is associated with elevated ROS [102, 103], and antioxidants in the DSP could inhibit such signalling. We obtained very similar reduction in growth rates (with no change in mature size) in normal C57/BL6-SJL mice fed our original complex supplement rich in antioxidants. In that case, there was a significant 11% extension in life span [16] but no reduction in food intake relative to untreated controls (P = 0.95, unpublished). The original DSP also increased motor activity of mice [17] that could also reduce growth efficiency. It is also worth mentioning that the maximal longevity of ~800 days in Trp53+/– mice that escaped cancer is not atypical of the C57BL/6 strain. [105]
The size of Trp53+/– mice approaches that of transgenic growth hormone mice (Tg). We previously obtained a statistically significant increase in longevity of Tg mice (27%) with our original DSP. [16] Various types of Tg also have increased susceptibility to cancer (Prof. A. Bartke, personal communication) as might be expected from the known impacts of growth hormone and IGF-1 on the TOR pathway and cancer. [49, 102, 103] Some increase in the survival of supplemented Tg likely reflected reduced cancers.
Clinical potential of the DSP
An 11-year study of treatment and follow-up for cancer in >14 000 physicians taking a daily multivitamin (Centrum Silver®) (versus placebo) found modest but significant reductions in total cancer with no indicators of significant harm. [13] This is one of the first clinical studies to experimentally address a truly broad-spectrum formulation. Overall, a 6.36% reduction in total cancer was observed. Excluding prostate cancer, the overall reduction was ~11%. For those with a baseline history of cancer, treatment reduced total cancer by 22.6%.
This study involved men 50 or older. Examination of cancer incidence with greater follow-up time showed progressively greater divergence (improved resolution) between treated and untreated groups for cumulative cancer and colorectal cancer (but not prostate cancer). Similar trends for some cancers were reported by others. [106, 107] Given the likely long-term cumulative benefits of supplements and prolonged age-related development of tumorigenesis, substantial monitoring and follow- up periods are required to detect cancer benefits in humans. [13, 108] Our lifetime study of mice is instructive in that expression of cancers and protection by the DSP were largely limited to advanced ages (Figure 1).
Many studies testing individual vitamins and some testing multivitamins have detected no benefit for cancer. [109] Alternatively, extensive evidence supports effectiveness of diverse phytochemicals on pathways relevant to aging and cancer. [110] Given the well-documented pro-oxidant capacity of many antioxidants when given individually in high doses, negative results can be a prophecy of design that for clinical trials seems ill advised. Many negative studies classified as ‘multivitamin’ test only 2–4 ingredients, so pro-oxidant risks likely remain. Prevalence of such studies and those of insufficient duration must strongly bias outcomes of meta-analyses where a truly effective formulation could be averaged out.
Despite considerable evidence against nutraceutical efficacy against cancer, other studies support the Centrum® multivitamin trial. [13] The ‘Linxian Chinese Cancer Prevention Trial’ found a 13% reduction in cancer mortality and a 9% reduction in total mortality for a supplement containing beta carotene, vitamin E and selenium. [106] Remarkably, benefits of multivitamin supplementation persisted for 10 years following completion of the trial, although this was largely limited to participants younger than 55 years. [111] A study testing a combination of vitamin C, vitamin E, beta carotene, selenium and zinc obtained significant benefit for cancer and total mortality limited to men (107). The ‘Nurses’ Health Study’ detected that prolonged use of multivitamins with folate ameliorated colon cancer but only in a 15-year follow up. [112] The ‘Cancer Prevention Study II’ found an inverse relationship between long-term multivitamin use with colon cancer and mortality. [108] A study of vitamin E and C, selenium and zinc intake in >23,000 participants who are 40–74 years old found that those with diets containing high amounts were 67% less likely to develop pancreatic cancer. [14] Another large study calculating the total antioxidant capacity of an Italian diet found a significant inverse relationship between colon cancer and dietary antioxidant capacity. [113] It must be emphasised that the DSP is an anti-cancer cocktail and not simply an antioxidant supplement. In this regard, the efficacy of aspirin in reducing diverse cancers (~20% reduction overall) is established for humans. [93, 94]
Our study differs from others in the larger number and higher dosages of ingredients employed, as well as a formulation designed to impact multiple mechanisms of aging and cancer (i.e. all supplements are not equal and the whole is greater than the sum of the parts). This may simultaneously obtain global alterations in redox and energy status and diverse impacts on cellular regulatory circuitry. This may be particularly relevant to cancers that exploit multiple pathways to evolve greater oncogenic potential and resist treatments.
Potential mechanisms of DSP action
Mechanisms explaining amelioration of cancer by the DSP include(i) reduction of ROS and prevention of genotoxic stress (suppressing initiation of cancer, regulatory distortion and metastatic evolution);
(ii) growth and cell cycle inhibition and
(iii) reducing signals inhibiting apoptosis that are generally over expressed in cancer.We have good support for the first. Although we found a small inhibition of growth, it seems insufficient to explain the degree of cancer reduction. Release of apoptosis by reducing the oxidative environment and enabling suppressed signalling systems is highly controversial.
Upregulation of TOR and growth by oxidative conditions can suppress FOXO. [102, 103] Many crucial enzymes involved in control of redox, repair, the cell cycle and apoptosis contain cysteine residues conveying redox regulation (including p53). [66] Thus, reducing the oxidative environment of cancer could inhibit IGF-1 signalling and TOR (and their associated survival signal) while upregulating pro-apoptotic FOXO. Several studies found that antioxidants (N-acetylcysteine, tempol, propyl gallate, dithiothreitol, melatonin) potently induce apoptosis in some cancers including breast cancer cells with breast cancer 1 (BRCA1) deficiency and colon cancer. [114, 115] Others argue, however, that effects of antioxidants reflect their pro-oxidant potential. [116] Increasing oxidative stress is also highlighted in chemotherapy. [117] The controversy particularly focuses on the Kelch like-ECH-associated protein 1 (KEAP1)/ nuclear factor 2 (NRF2) stress response system. Although FOXO may be inhibited in cancer, NRF2 can be activated to potentially protect cancers from excessive oxidative stress and chemotherapy. [117]
Our DSP was designed to ensure recycling among antioxidants, and we documented that our original formulation has antioxidant rather than any pro-oxidant properties. [17, 19, 58, 75] We are currently examining apoptosis in cancers found in DSP-treated mice to determine if cell killing might be achieved by reducing the oxidative conditions supporting cancer or by otherwise supporting FOXO activity.
Some suggest that phytochemicals and antioxidants have no anti-aging or cancer benefits. Our results certainly do not support Watson’s 2013 [12] claim that antioxidants cause cancer, although it is likely that the DSP could impede chemotherapy agents that act via elevated oxidative stress. The real question is whether the DSP itself is a novel form of chemotherapy.
Funding
C.D.R. was supported by a grant from the Natural Sciences and Engineering Research Council of Canada and D.B. was supported by a grant from the US Department of Energy Low Dose Radiation Research Program (grant number DE-FG02-07ER64343).
Acknowledgements
We thank Zoya Tov for assisting in preparation of the dietary supplement, Veterinary Pathologist Dr Dean Percy for carrying out the histopathological examinations, Prof. Joao Pedro de Magalhaes for helpful suggestions in review and our editor Dr Charles Limoli.
REFERENCES
Levine, A. J. (1997) p53, the cellular gatekeeper for growth and division. Cell, 88, 323–331.
Venkatachalam, S., Tyner, S. D., Pickering, C. R., Boley, S., Recio, L., French, J. E. and Donehower, L. A. (2001) Is p53 haploinsufficient for tumor suppression? Implications for the p53+/- mouse model in carcinogenicity testing. Toxicol. Pathol., 29 Suppl, 147–154.
Rodier, F., Campisi, J. and Bhaumik, D. (2007) Two faces of p53: aging and tumor suppression. Nucleic Acids Res., 35, 7475–7484.
Hollstein, M., Rice, K., Greenblatt, M. S., et al. (1994) Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res., 22, 3551–3555.
Greenblat, M. S., Bennet, W. P., Hollstein, M. and Harris, C. C. (1994) Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res., 54, 4855–4878.
Vousden, K. H. and Lane, D. P. (2007) p53 in health and disease. Nat. Rev. Mol. Cell Biol., 8, 275–283.
Tyner, S. D., Venkatachalam, S., Choi, J., et al. (2002) p53 mutant mice that display early ageing-associated phenotypes. Nature, 415, 45–53.
Reinhardt, H. C. and Schumacher, B. (2012) The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet., 28, 128–136.
World Health Organization (2013) Cancer Facts and Figures. World Health Organization, Regional Office for Europe, Denmark.
Siegel, R., Naishadham, D. and Jemal, A. (2012) Cancer statistics, 2012. CA. Cancer J. Clin., 62, 10–29.
Bast, A. and Haenen, G. R. (2013) Ten misconceptions about antioxidants. Trends Pharmacol. Sci., 34, 430–436.
Watson, J. (2013) Oxidants, antioxidants and the current incurability of metastatic cancers. Open Biol., 3, 120144. doi:10.1098/rsob.120144.
Gaziano, J. M., Sesso, H. D., Christen, W. G., et al. (2012) Multivitamins in the prevention of cancer in men: the Physicians’ Health Study II randomized controlled trial. JAMA, 308, 1871–1880.
Banim, P. J., Luben, R., McTaggart, A., Welch, A., Wareham, N., Khaw, K. T. and Hart, A. R. (2013) Dietary antioxidants and the aetiology of pancreatic cancer: a cohort study using data from food diaries and biomarkers. Gut, 62, 1489–1496.
Lemon, J. A., Boreham, D. R. and Rollo, C. D. (2003) A dietary supplement abolishes age-related cognitive decline in transgenic mice expressing elevated free radical processes. Exp. Biol. Med. (Maywood), 228, 800–810.
Lemon, J. A., Boreham, D. R. and Rollo, C. D. (2005) A complex dietary supplement extends longevity of mice. J. Gerontol. A. Biol. Sci. Med. Sci., 60, 275–279.
Aksenov, V., Long, J., Lokuge, S., Foster, J. A., Liu, J. and Rollo, C. D. (2010) Dietary amelioration of locomotor, neurotransmitter and mitochondrial aging. Exp. Biol. Med. (Maywood), 235, 66–76.
Aksenov, V., Long, J., Liu, J., Szechtman, H., Khanna, P., Matravadia, S. and Rollo, C. D. (2013) A complex dietary supplement augments spatial learning, brain mass, and mitochondrial electron transport chain activity in aging mice. Age (Dordr), 35, 23–33.
Long, J., Aksenov, V., Rollo, C. D. and Liu, J. (2012) A complex dietary supplement modulates nitrative stress in normal mice and in a new mouse model of nitrative stress and cognitive aging. Mech. Ageing Dev., 133, 523–529.
Cho, W. C. S. (ed.) (2013) Cancer Chemoprevention and Treatment by Diet Therapy. Evidence-based Anticancer Complementary and Alternative Medicine. Vol. 5. Springer Science Business Media, Dordrechtpy.
Banerjee, S., Azmi, A., Bao, B. and Sarkar, F. H. (2013) Attenuation of multifocal cell survival signaling by bioactive phytochemicals in the prevention and therapy of cancer. In Cho, W. C. S. (ed.), Cancer Chemoprevention and Treatment by Diet Therapy. Evidence-based Anticancer Complementary and Alternative Medicine. Vol. 5. Springer Science Business Media, Dordrechtpy, pp. 269–310.
Lin, S. S., Lai, K. C., Hsu, S. C., Yang, J. S., Kuo, C. L., Lin, J. P., Ma, Y. S., Wu, C. C. and Chung, J. G. (2009) Curcumin inhibits the migration and invasion of human A549 lung cancer cells through the inhibition of matrix metalloproteinase-2 and -9 and vascular endothelial growth factor (VEGF). Cancer Lett., 285, 127–133.
Kunnumakkara, A. B., Anand, P. and Aggarwal, B. B. (2008) Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett., 269, 199–225.
Liontas, A. and Yeger, H. (2004) Curcumin and resveratrol induce apoptosis and nuclear translocation and activation of p53 in human neuroblastoma. Anticancer Res., 24, 987–998.
Shishodia, S., Amin, H. M., Lai, R. and Aggarwal, B. B. (2005) Curcumin (diferuloylmethane) inhibits constitutive NF-kappaB activation, induces G1/S arrest, suppresses proliferation, and induces apoptosis in mantle cell lymphoma. Biochem. Pharmacol., 70, 700–713.
Hatcher, H. C., Torti, F. M. and Torti, S. V. (2012) Chapter 12: Curcumin, oxidative stress, and cancer therapy. In Spitz, D. R., Dornfeld, K. J., Krishnan, K. and Gius, D. (eds), Oxidative Stress in Cancer Biology and Therapy, Oxidative Stress in Applied Basic Research and Clinical Practice. Springer Science+Business Media, LLC, New York, pp. 233– 256. doi:10.1007/978-1-61779-397-4_12
Kotha, A., Sekharam, M., Cilenti, L., Siddiquee, K., Khaled, A., Zervos, A. S., Carter, B., Turkson, J. and Jove, R. (2006) Resveratrol inhibits Src and Stat3 signaling and induces the apoptosis of malignant cells containing activated Stat3 protein. Mol. Cancer Ther., 5, 621–629.
Gatouillat, G., Balasse, E., Joseph-Pietras, D., Morjani, H. and Madoulet, C. (2010) Resveratrol induces cell-cycle disruption and apoptosis in chemoresistant B16 melanoma. J. Cell. Biochem., 110, 893–902.
Shukla, Y. and Singh, R. (2011) Resveratrol and cellular mechanisms of cancer prevention. Ann. N. Y. Acad. Sci., 1215, 1–8.
Zheng, J., Chen, L. L., Zhang, H. H., Hu, X., Kong, W. and Hu, D. (2012) Resveratrol improves insulin resistance of catch-up growth by increasing mitochondrial complexes and antioxidant function in skeletal muscle. Metabolism, 61, 954–965.
Rao, A. V. and Agarwal, S. (2000) Role of antioxidant lycopene in cancer and heart disease. J. Am. Coll. Nutr., 19, 563–569.
Teodoro, A. J., Oliveira, F. L., Martins, N. B., Maia, G. de. A., Martucci, R. B. and Borojevic, R. (2012) Effect of lycopene on cell viability and cell cycle progression in human cancer cell lines. Cancer Cell Int., 12, 36.
Levy, J., Bosin, E., Feldman, B., Giat, Y., Miinster, A., Danilenko, M. and Sharoni, Y. (1995) Lycopene is a more potent inhibitor of human cancer cell proliferation than either alpha-carotene or beta-carotene. Nutr. Cancer, 24, 257–266.
Jeong, J. H., An, J. Y., Kwon, Y. T., Rhee, J. G. and Lee, Y. J. (2009) Effects of low dose quercetin: cancer cell-specific inhibition of cell cycle progression. J. Cell. Biochem., 106, 73–82.
Sung, M. S., Lee, E. G., Jeon, H. S., Chae, H. J., Park, S. J., Lee, Y. C. and Yoo, W. H. (2012) Quercetin inhibits IL-1β-induced proliferation and production of MMPs, COX-2, and PGE2 by rheumatoid synovial fibroblast. Inflammation, 35, 1585–1594.
Zhang, M., Swarts, S. G., Yin, L., et al. (2011) Antioxidant properties of quercetin. Adv. Exp. Med. Biol., 701, 283–289.
Huang, C. Y., Chan, C. Y., Chou, I. T., Lien, C. H., Hung, H. C. and Lee, M. F. (2013) Quercetin induces growth arrest through activation of FOXO1 transcription factor in EGFR-overexpressing oral cancer cells. J. Nutr. Biochem., 24, 1596–1603.
Koyama, S., Cobb, L. J., Mehta, H. H., Seeram, N. P., Heber, D., Pantuck, A. J. and Cohen, P. (2010) Pomegranate extract induces apoptosis in human prostate cancer cells by modulation of the IGF-IGFBP axis. Growth Horm. IGF Res., 20, 55–62.
Banerjee, N., Talcott, S., Safe, S. and Mertens-Talcott, S. U. (2012) Cytotoxicity of pomegranate polyphenolics in breast cancer cells in vitro and vivo: potential role of miRNA-27a and miRNA-155 in cell survival and inflammation. Breast Cancer Res. Treat., 136, 21–34.
El Kar, C., Ferchichi, A., Attia, F. and Bouajila, J. (2011) Pomegranate (Punica granatum) juices: chemical composition, micronutrient cations, and antioxidant capacity. J. Food Sci., 76, C795–C800.
Mitchel, R. E., Burchart, P. and Wyatt, H. (2007) Fractionated, lowdose- rate ionizing radiation exposure and chronic ulcerative dermatitis in normal and Trp53 heterozygous C57BL/6 mice. Radiat. Res., 168, 716–724.
Landi, M. T., Chatterjee, N., Yu, K., et al. (2009) A genome-wide association study of lung cancer identifies a region of chromosome 5p15 associated with risk for adenocarcinoma. Am. J. Hum. Genet., 85, 679–691.
Bogenrieder, T. and Herlyn, M. (2003) Axis of evil: molecular mechanisms of cancer metastasis. Oncogene, 22, 6524–6536.
Chambers, A. F., Groom, A. C. and MacDonald, I. C. (2002) Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer, 2, 563–572.
Hanahan, D. and Weinberg, R. A. (2000) The hallmarks of cancer. Cell, 100, 57–70.
Cui, X. (2012) Reactive oxygen species: the Achilles’ heel of cancer cells? Antioxid. Redox Signal., 16, 1212–1214.
Slomiany, M. G., Black, L. A., Kibbey, M. M., Day, T. A. and Rosenzweig, S. A. (2006) IGF-1 induced vascular endothelial growth factor secretion in head and neck squamous cell carcinoma. Biochem. Biophys. Res. Commun., 342, 851–858.
Zhang, M., Zhou, Q., Liang, Q. Q., et al. (2009) IGF-1 regulation of type II collagen and MMP-13 expression in rat endplate chondrocytes via distinct signaling pathways. Osteoarthritis Cartilage, 17, 100–106.
Patel, A. C., Nunez, N. P., Perkins, S. N., Barrett, J. C. and Hursting, S. D. (2004) Effects of energy balance on cancer in genetically altered mice. J. Nutr., 134, 3394S–3398S.
Reuter, S., Gupta, S. C., Chaturvedi, M. M. and Aggarwal, B. B. (2010) Oxidative stress, inflammation, and cancer: how are they linked? Free Radic. Biol. Med., 49, 1603–1616.
Munaut, C., Noël, A., Hougrand, O., Foidart, J. M., Boniver, J. and Deprez, M. (2003) Vascular endothelial growth factor expression correlates with matrix metalloproteinases MT1-MMP, MMP-2 and MMP-9 in human glioblastomas. Int. J. Cancer, 106, 848–855.
Mori, K., Shibanuma, M. and Nose, K. (2004) Invasive potential induced under long-term oxidative stress in mammary epithelial cells. Cancer Res., 64, 7464–7472.
Ushio-Fukai, M. and Alexander, R. W. (2004) Reactive oxygen species as mediators of angiogenesis signaling: role of NAD(P)H oxidase. Mol. Cell. Biochem., 264, 85–97.
Ries, C. and Petrides, P. E. (1995) Cytokine regulation of matrix metalloproteinase activity and its regulatory dysfunction in disease. Biol. Chem. Hoppe. Seyler., 376, 345–355.
Bollrath, J. and Greten, F. R. (2009) IKK/NF-kappaB and STAT3 pathways: central signalling hubs in inflammation-mediated tumour promotion and metastasis. EMBO Rep., 10, 1314–1319.
Ristimäki, A., Narko, K., Enholm, B., Joukov, V. and Alitalo, K. (1998) Proinflammatory cytokines regulate expression of the lymphatic endothelial mitogen vascular endothelial growth factor-C. J. Biol. Chem., 273, 8413–8418.
Greenlee, R. T., Hill-Harmon, M. B., Murray, T. and Thun, M. (2001) Cancer statistics, 2001. CA. Cancer J. Clin., 51, 15–36.
Lemon, J. A., Rollo, C. D. and Boreham, D. R. (2008) Elevated DNA damage in a mouse model of oxidative stress: impacts of ionizing radiation and a protective dietary supplement. Mutagenesis, 23, 473–482.
Sablina, A. A., Budanov, A. V., Ilyinskaya, G. V., Agapova, L. S., Kravchenko, J. E. and Chumakov, P. M. (2005) The antioxidant function of the p53 tumor suppressor. Nat. Med., 11, 1306–1313.
Algra, A. M. and Rothwell, P. M. (2012) Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol., 13, 518–527.
Chan, W. S., Page, C. M., Maclellan, J. R. and Turner, G. A. (1988) The growth and metastasis of four commonly used tumour lines implanted into eight different sites: evidence for site and tumour effects. Clin. Exp. Metastasis, 6, 233–244.
de Magalhăes, J. P. (2013) How ageing processes influence cancer. Nat. Rev. Cancer, 13, 357–365.
Valko, M., Rhodes, C. J., Moncol, J., Izakovic, M. and Mazur, M. (2006) Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact., 160, 1–40.
Waris, G. and Ahsan, H. (2006) Reactive oxygen species: role in the development of cancer and various chronic conditions. J. Carcinog., 5, 14.
Pignatelli, B., Li, C. Q., Boffetta, P., et al. (2001) Nitrated and oxidized plasma proteins in smokers and lung cancer patients. Cancer Res., 61, 778–784.
Caputo, F., Vegliante, R. and Ghibelli, L. (2012) Redox modulation of the DNA damage response. Biochem. Pharmacol., 84, 1292–1306.
Seo, Y. R. and Jung, H. J. (2004) The potential roles of p53 tumor suppressor in nucleotide excision repair (NER) and base excision repair (BER). Exp. Mol. Med., 36, 505–509.
Serafini, M., Bellocco, R., Wolk, A. and Ekström, A. M. (2002) Total antioxidant potential of fruit and vegetables and risk of gastric cancer. Gastroenterology, 123, 985–991.
Aggarwal, B. B., Bhardwaj, A., Aggarwal, R. S., Seeram, N. P., Shishodia, S. and Takada, Y. (2004) Role of resveratrol in prevention and therapy of cancer: preclinical and clinical studies. Anticancer Res., 24, 2783–2840.
Van Remmen, H., Ikeno, Y., Hamilton, M., et al. (2003) Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol. Genomics, 16, 29–37.
Oberley, T. D. and Oberley, L. W. (1997) Antioxidant enzyme levels in cancer. Histol. Histopathol., 12, 525–535.
Yang, J., Lam, E. W., Hammad, H. M., Oberley, T. D. and Oberley, L. W. (2002) Antioxidant enzyme levels in oral squamous cell carcinoma and normal human oral epithelium. J. Oral Pathol. Med., 31, 71–77.
Goh, J., Enns, L., Fatemie, S., Hopkins, H., Morton, J., Pettan-Brewer, C. and Ladiges, W. (2011) Mitochondrial targeted catalase suppresses invasive breast cancer in mice. BMC Cancer, 11, 191.
Bonassi, S., Hagmar, L., Strömberg, U., et al. (2000) Chromosomal aberrations in lymphocytes predict human cancer independently of exposure to carcinogens. European Study Group on Cytogenetic Biomarkers and Health. Cancer Res., 60, 1619–1625.
Lemon, J. A., Rollo, C. D., McFarlane, N. M. and Boreham, D. R. (2008) Radiation-induced apoptosis in mouse lymphocytes is modified by a complex dietary supplement: the effect of genotype and gender. Mutagenesis, 23, 465–472.
Ribeiro, M. L., Arçari, D. P., Squassoni, A. C. and Pedrazzoli, J., Jr. (2007) Effects of multivitamin supplementation on DNA damage in lymphocytes from elderly volunteers. Mech. Ageing Dev., 128, 577–580.
Rollo, C. D. (2007) Multidisciplinary aspects of regulatory systems relevant to multiple stressors: aging, xenobiotics and radiation. In Mothersill, C., Mosse, I. and Seymour, C. (eds), Multiple Stressors: a Challenge for the Future. Springer (NATO Science), Dordrecht, The Netherlands, pp. 185–224.
Rollo, C. D. (2009) Dopamine and aging: intersecting facets. Neurochem. Res., 34, 601–629.
Raha, S. and Robinson, B. H. (2000) Mitochondria, oxygen free radicals, disease and aging. Trends Biochem Sci., 25, 502–508.
Bowling, A. C., Mutisya, E. M., Walker, L. C., Price, D. L., Cork, L. C. and Beal, M. F. (1993) Age-dependent impairment of mitochondrial function in primate brain. J. Neurochem., 60, 1964–1967.
Kwong, L. K. and Sohal, R. S. (2000) Age-related changes in activities of mitochondrial electron transport complexes in various tissues of the mouse. Arch. Biochem. Biophys., 373, 16–22.
Merry, B. J. (2004) Oxidative stress and mitochondrial function with aging–the effects of calorie restriction. Aging Cell, 3, 7–12.
Guarente, L. (2008) Mitochondria–a nexus for aging, calorie restriction, and sirtuins? Cell, 132, 171–176.
Barja, G. (2004) Free radicals and aging. Trends Neurosci., 27, 595–600.
Albanes, D. (1987) Total calories, body weight, and tumor incidence in mice. Cancer Res., 47, 1987–1992.
Nemat, K., Yadollah, A. and Mahdi, M. (2009) Chronic inflammation and oxidative stress as a major cause of age-related diseases and cancer. Recent Pat. Inflamm. Allergy Drug Discov., 3, 73–80.
Grivennikov, S. I., Greten, F. R. and Karin, M. (2010) Immunity, inflammation, and cancer. Cell, 140, 883–899.
Ohshima, H., Tatemichi, M. and Sawa, T. (2003) Chemical basis of inflammation-induced carcinogenesis. Arch. Biochem. Biophys., 417, 3–11.
Dröge, W. (2002) Free radicals in the physiology control of cell function. Physiol. Rev., 82, 47–95.
Bonizzi, G., Piette, J., Merville, M. O. and Bours, V. (2000) Cell typespecific role of reactive oxygen species in nuclear factor κB activation by interleukin-1. Biochem. Pharmacol., 59, 7–11.
Langman, M. J., Cheng, K. K., Gilman, E. A. and Lancashire, R. J. (2000) Effect of anti-inflammatory drugs on overall risk of common cancer: case-control study in general practice research database. BMJ, 320, 1642–1646.
Kawamori, T., Lubet, R., Steele, V. E., Kelloff, G. J., Kaskey, R. B., Rao, C. V. and Reddy, B. S. (1999) Chemopreventive effect of curcumin, a naturally occurring anti-inflammatory agent, during the promotion/progression stages of colon cancer. Cancer Res., 59, 597–601.
Rothwell, P. M., Fowkes, F. G., Belch, J. F., Ogawa, H., Warlow, C. P. and Meade, T. W. (2011) Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet, 377, 31–41.
Rothwell, P. M., Price, J. F., Fowkes, F. G., et al. (2012) Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet, 379, 1602–1612.
Ryan, A. S. (2000) Insulin resistance with aging: effects of diet and exercise. Sports Med., 30, 327–346.
Fink, R. I., Kolterman, O. G., Griffin, J. and Olefsky, J. M. (1983) Mechanisms of insulin resistance in aging. J. Clin. Invest., 71, 1523–1535.
Kaaks, R. and Lukanova, A. (2001) Energy balance and cancer: the role of insulin and insulin-like growth factor-I. Proc. Nutr. Soc., 60, 91–106.
Boyd, D. B. (2003) Insulin and cancer. Integr. Cancer Ther., 2, 315–329.
Tsugane, S. and Inoue, M. (2010) Insulin resistance and cancer: epidemiological evidence. Cancer Sci., 101, 1073–1079.
Gonzalez-Angulo, A. M. and Meric-Bernstam, F. (2010) Metformin: a therapeutic opportunity in breast cancer. Clin. Cancer Res., 16, 1695–1700.
Price, N. L., Gomes, A. P., Ling, A. J., et al. (2012) SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab., 15, 675–690.
Rollo, C. D. (2010) Aging and the mammalian regulatory triumvirate. Aging Dis., 1, 105–138.
Rollo, C. D. (2012) Circadian redox regulation. In Pantopoulos, K. and Shipper, H. M. (eds), Principles of Free Radical Biomedicine. Nova Science Publisher, New York, pp. 575–627.
Maier, B., Gluba, W., Bernier, B., Turner, T., Mohammad, K., Guise, T., Sutherland, A., Thorner, M. and Scrable, H. (2004) Modulation of mammalian life span by the short isoform of p53. Genes Dev., 18, 306–319.
Sohal, R. S., Agarwal, S., Candas, M., Forster, M. J. and Lal, H. (1994) Effect of age and caloric restriction on DNA oxidative damage in different tissues of C57BL/6 mice. Mech. Ageing Dev., 76, 215–224.
Blot, W. J., Li, J. Y., Taylor, P. R., et al. (1993) Nutrition intervention trials in Linxian, China: supplementation with specific vitamin/mineral combinations, cancer incidence, and disease-specific mortality in the general population. J. Natl. Cancer Inst., 85, 1483–1492.
Hercberg, S., Galan, P., Preziosi, P., Bertrais, S., Mennen, L., Malvy, D., Roussel, A. M., Favier, A. and Briançon, S. (2004) The SU.VI.MAX Study: a randomized, placebo-controlled trial of the health effects of antioxidant vitamins and minerals. Arch. Intern. Med., 164, 2335–2342.
Jacobs, E. J., Connell, C. J., Chao, A., McCullough, M. L., Rodriguez, C., Thun, M. J. and Calle, E. E. (2003) Multivitamin use and colorectal cancer incidence in a US cohort: does timing matter? Am. J. Epidemiol., 158, 621–628.
Reid, M. and Marshall, J. (2013) Chemoprevention of cancer: from nutritional epidemiology to clinical trials. In Miller, A. B. (ed.), Epidemiologic Studies in Cancer Prevention and Screening, Statistics for Biology and Health. Springer Science Business Media, New York, pp. 141–162.
Banerjee, S. and Rajamani, P. (2013) Cellular, molecular, and biological perspective of polyphenols in chemoprevention and therapeutic adjunct in cancer. In Ramawat, K. G. and Merillon, J. M. (eds), Natural Products. Springer-Verlag, Berlin, Heidelberg, pp. 2175–2253.
Qiao, Y. L., Dawsey, S. M., Kamangar, F., et al. (2009) Total and cancer mortality after supplementation with vitamins and minerals: follow-up of the Linxian General Population Nutrition Intervention Trial. J. Natl. Cancer Inst., 101, 507–518.
Giovannucci, E., Stampfer, M. J., Colditz, G. A., Hunter, D. J., Fuchs, C., Rosner, B. A., Speizer, F. E. and Willett, W. C. (1998) Multivitamin use, folate, and colon cancer in women in the nurses’ health study. Ann. Intern. Med., 129, 517–524.
La Vecchia, C., Decarli, A., Serafini, M., et al. (2013) Dietary total antioxidant capacity and colorectal cancer: a large case-control study in Italy. Int. J. Cancer, 133, 1447–1451.
Chang, Y. J., Huang, Y. P., Li, Z. L. and Chen, C. H. (2012) GRP78 Knockdown enhances apoptosis via the down-regulation of oxidative stress and Akt pathway after epirubicin treatment in colon cancer DLD-1 cells. PLoS ONE, 7, e35123.
Martinez-Outschoorn, U. E., Balliet, R., Lin, Z., et al. (2012) BRCA1 mutations drive oxidative stress and glycolysis in the tumor microenvironment: implications for breast cancer prevention with antioxidant therapies. Cell Cycle, 11, 4402–4413.
Azmi, A. S., Sarkar, F. H. and Hadi, S. (2013) Pro-oxidant activity of dietary chemopreventive agents: an under-appreciated anti-cancer property. F1000Res., 2, 135.
Sporn, M. B. and Liby, K. T. (2012) NRF2 and cancer: the good, the bad and the importance of context. Nat. Rev. Cancer, 12, 564–571.

Return to NUTRITION
Since 8–06-2018


| Home Page | Visit Our Sponsors | Become a Sponsor |
Please read our DISCLAIMER |